14.5.1.1. Spatially Structured Molecule Description¶
Warning
The spatially structured molecule interface is relatively new and subject to change.
Spatially Structured Molecules can be:
Defined in CellBlender
Combined as complexes in CellBlender
Exported for simulation in MCell

14.5.1.1.1. Molecules and Components¶
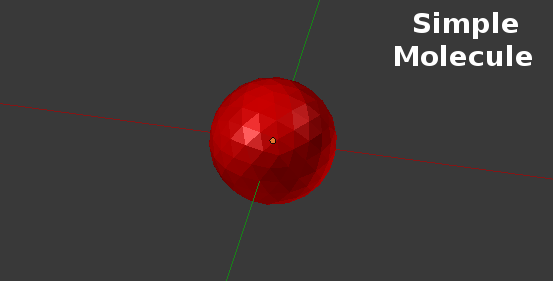
Using CellBlender’s Spatially Structured Molecules, the geometry within a molecule is specified relative to the molecule’s “center”. The “center” is an arbitrary location within a simple molecule and has traditionally been represented with a “glyph” of some sort:
With Spatially Structured Molecules, this “center” is augmented with a number of component locations that act as binding sites for other molecules:
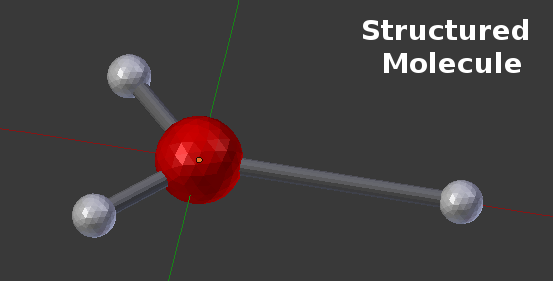
This example shows a “center” molecule surrounded by 3 different “components” (also known as binding sites). This particular Molecule was defined in CellBlender with the “Molecule Structure” panel as shown here:
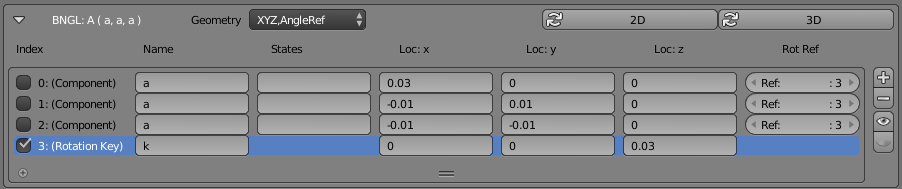
As you can see from the panel, this molecule contains three items listed as “Components” and one item listed as a “Rotation Key”. The 3 components (all named “a”) are the standard BNGL components and they are given spatial positions in the table (“Loc:x”, “Loc:y”, “Loc:z”). You’ll notice that the first component in the list shows a “Loc:x” value of 0.03 compared to -0.01 for the other “Loc:x” values. That’s the component (smaller white sphere) to the far right in the picture. Similarly, the other two components (indexes 1 and 2) show negative x values and y values of opposite sign. Those are the two components (white spheres) to the left in the picture. The last component (Index=3, Name=”k”) isn’t shown in this simple picture, and it’s not a true “binding component”. It’s used as a reference “key” for the rotation axes of other binding components. This is shown more clearly in this picture:
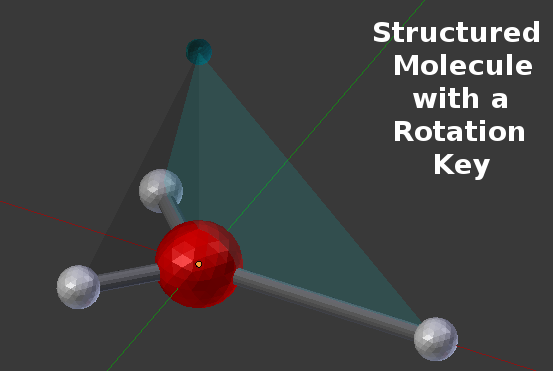
This picture shows the 4th item (a “Rotation Key”) along with the three Rotation Alignment Planes that it creates with each binding component. You’ll notice that the “Rot Ref” column of the previous table shows a “Ref: 3” for the first three items. That means that they are each using the “Rotation Key” at index 3 to define their rotational orientation with respect to any binding molecules. That rotation key is at a coordinate location of (0,0,0.03) which places it directly above the origin along the Z axis. This is a particularly convenient rotation key to use for molecules that are laid out in the X-Y plane (as in this example).
14.5.1.1.2. Binding of Molecules via Components¶
The following picture shows one example of how two of these molecules might be joined to form the BNGL product of A(a!1).A(a!1):
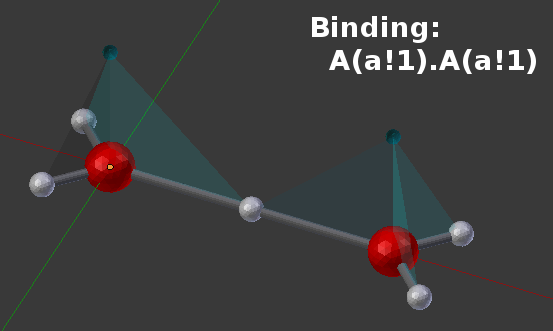
Note that because all of the components are named “a”, there are a number of physical configurations that might represent that same BNGL product:
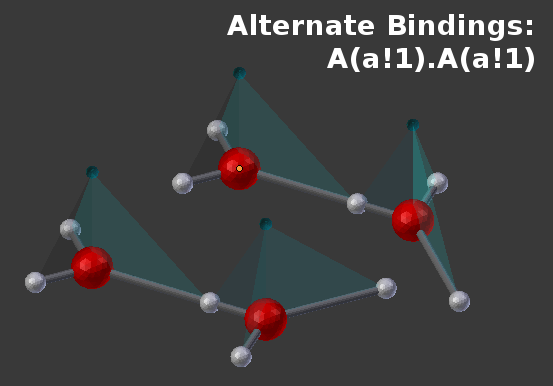
In cases like this (where different components have different physical locations), it may be desirable to give those components different names so they can be used differently in the rules that define the products.
14.5.1.1.3. Binding Angles and Rotation Reference Keys¶
While every component that is intended for binding should have a reference plane, the following picture shows the earlier A(a!1).A(a!1) complex with only the bound reference planes visible:
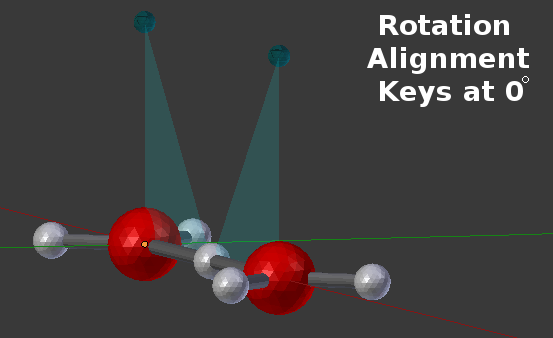
In that example, the two reference planes are lined up at 0 degrees of rotation. However, bonds can also be made at arbitrary angles between the reference planes as shown in this example where the bonding is specified to be at a 45 degree angle:
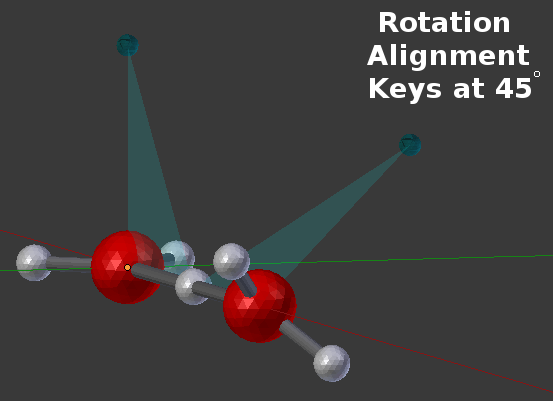
Reference keys (as shown above) can be very handy and are sometimes the simplest way to show how Rotation Alignment Planes can be defined. But many times, there’s no need to specify a special “rotation angle key” because one or more of the other components can easily fill that role. In that case, each component can simply reference another existing component to use as its rotation reference key. That’s shown in the following CellBlender panel where the “Rot Ref” column specifies other existing BNGL components and no special reference “key” is defined:
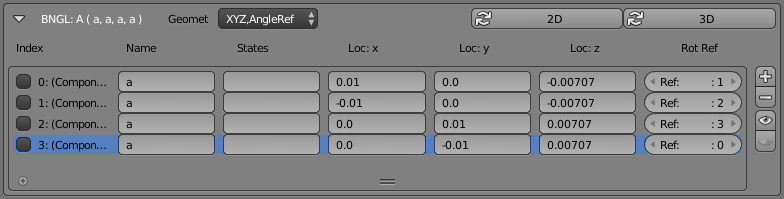
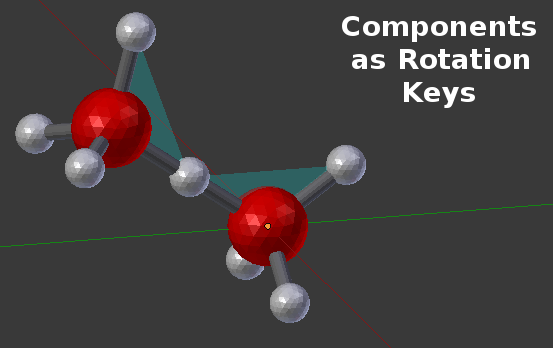
This panel still shows 4 items, but all 4 are real components. In this case, all of the components have been arranged as the vertices of a tetrahedron, and each component simply references another component as its binding angle key. Here’s an example built from two of those “A” molecules:
14.5.1.1.4. MCellR Simulations¶
Once the spatial structures have been defined for all molecules, the model can be simulated in an appropriate spatial simulator such as MCellR. The following image shows a simple LR model built and run in CellBlender with MCellR:
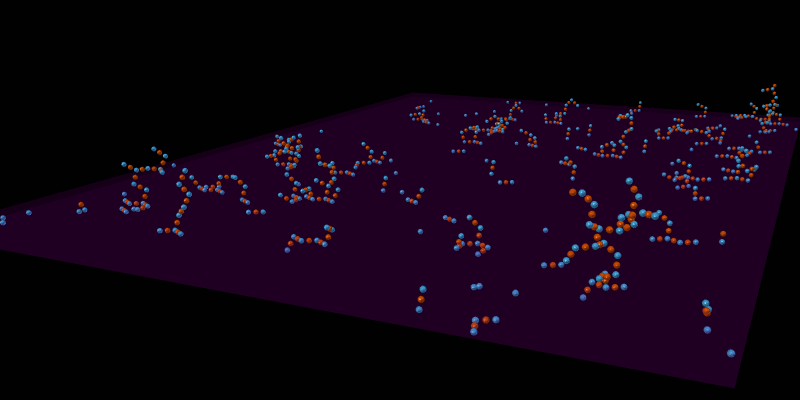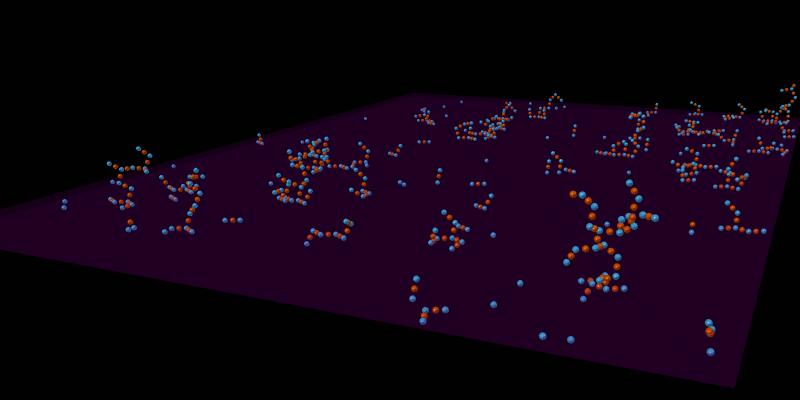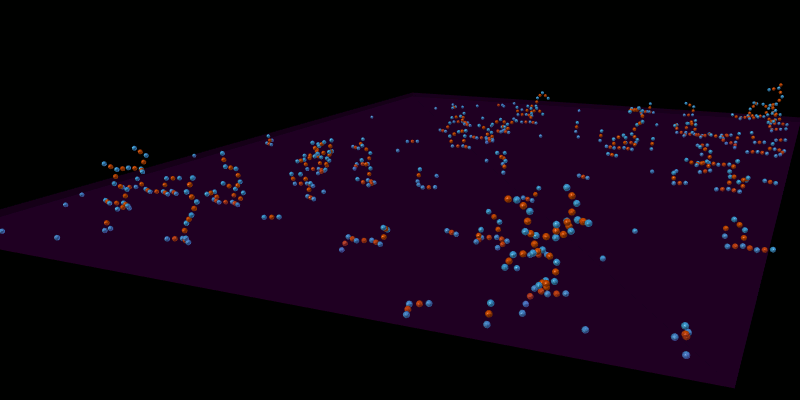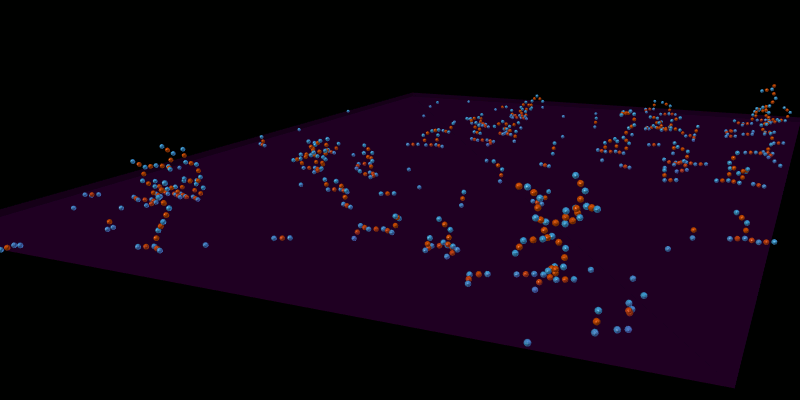
The next picture shows just one of the molecules that emerged from the simulation in the previous picture:
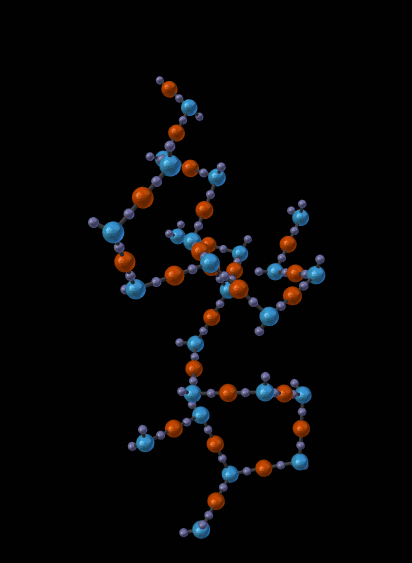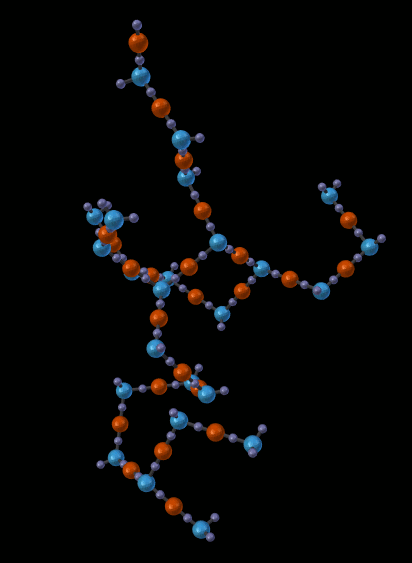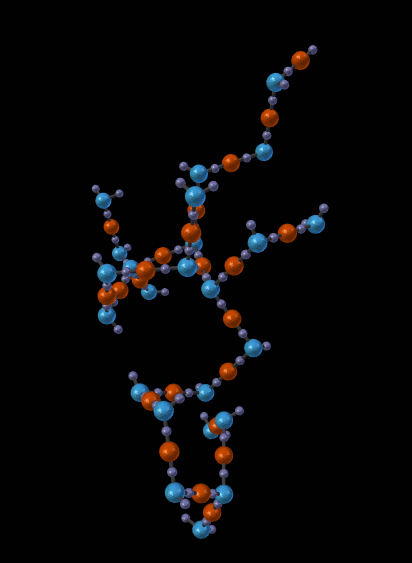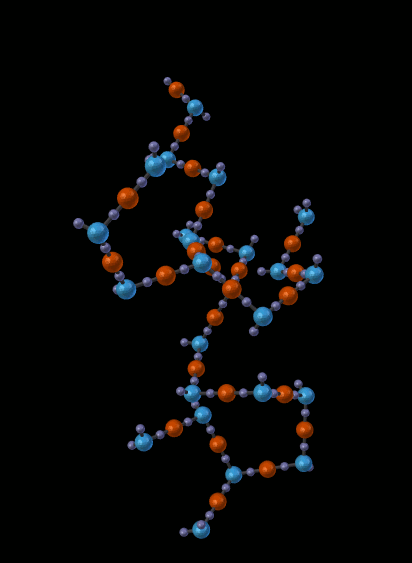
14.5.1.1.5. Other Features¶
CellBlender’s Molecule tools contain a number of helpful features for defining spatially structured molecules. For example, there are tools to automatically place components in either a 2D or 3D distribution around a molecule:
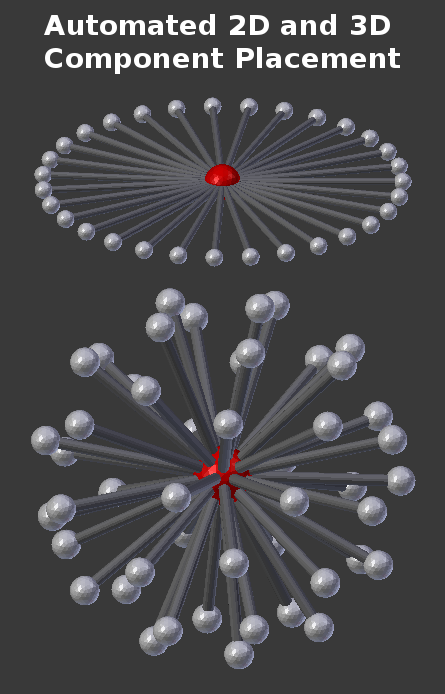
There are other tools to quickly visualize the locations of components on either single molecules or bonded molecules in a complex. For example, the following CellBlender Molecule subpanel was used to construct all of the examples in this tutorial:
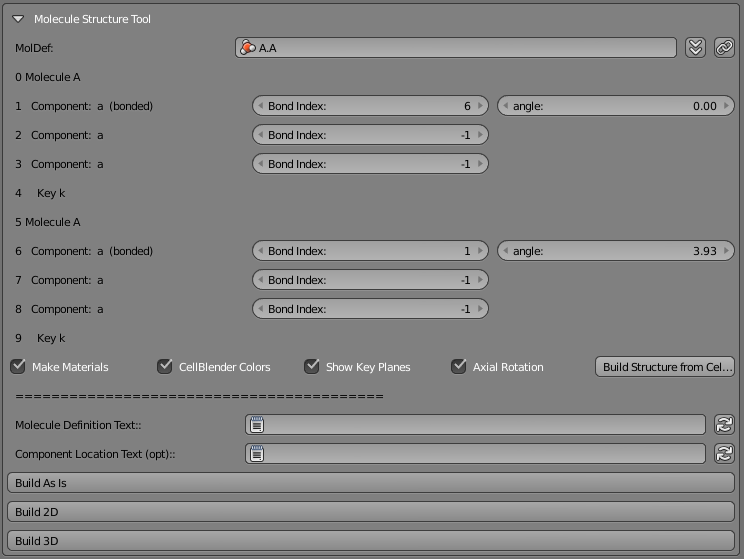
14.5.1.1.6. Summary¶
CellBlender’s Spatially Structured Molecule tools support flexible spatial structuring of complex molecules defined through the BioNetGen Language (BNGL) syntax.