14.5.1.2. Spatially Structured Tutorial (Construction)¶
Warning
The spatially structured molecule interface is relatively new and subject to change.
This tutorial will walk you through the steps of creating spatially structured molecules and manually binding them together to form spatially structured complexes. This tutorial assumes that you’re fairly familiar with Blender and CellBlender. It will use relatively brief instructions based on that assumption.

Start with Blender open. Enable CellBlender and Initialize CellBlender. Start a new project and save it to a “.blend” file. Then remove all objects from the scene.
Open the CellBlender “Settings & Preferences” panel, and enable the “BioNetGen Language Mode” option. The BioNetGen mode allows molecules to have components which are the binding points for building spatially structured complexes.
Then open the CellBlender “Molecules” panel and add a molecule named “A”. Then open the BNGL subpanel within the Molecules panel. Ensure that the panel is wide enough to see the Geometry drop down options. Then select the “XYZ,RotRef” Geometry option to begin building 3D Molecules.
We will start with a few single molecules with components and then work our way up to some interesting complexes.
Start by clicking the “+” button (in the BNGL panel) 4 times to add 4 components. The new components will each typically have default names like “C#”. These will be fine for now. Each component will also have an index number associated with it (in the “Index” column on the far left). In this tutorial, we will generally refer to each component using this index number.
Click the checkbox to the left of the last component (index number 3). This will change its type from being a “Component” to being a “Rotation Key”. We will want each of our other 3 components to use this as its rotation key, so enter the number “3” in the “Rot Ref” column for each of the other components (the column to the far right). Since there is only one rotation reference key in this molecule, you can also set this key for all components by clicking the “key” button that appears to the right.
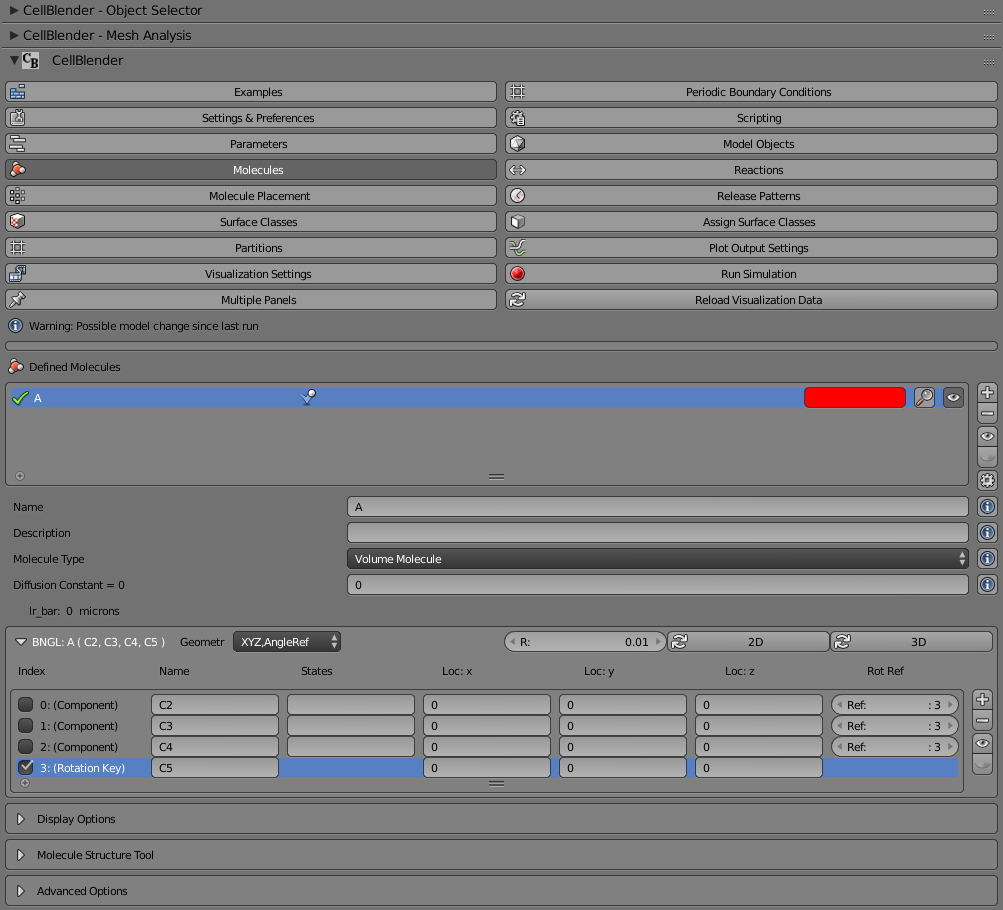
At this point, we have an “A” molecule with 3 components and 1 rotation reference key. This molecule could be used in a simulation, but all of its components are still at the coordinate (0,0,0) with respect to the molecule itself. If we knew the actual coordinates of these components, we could enter them in the “Loc” columns for x, y, and z. We could also start with some pre-defined locations using some of the built in tools.
For this example, let’s start by clicking the “2D” button. This will arrange our components equally around a radius “R” (specified just to the left of the “2D” button). you’ll notice that numbers are placed in both the x and y columns, but not in the z column. That’s appropriate for a 2D molecule. Note that it did not make any changes to the Rotation Key (component 3). You’ll have to set that yourself. For a 2D molecule, the z axis makes an ideal rotation reference key, so enter the number “0.03” in the “Loc: z” column of the last component.
To get a quick view of this arrangement, start by hiding all of your regular CellBlender molecules. Use the “grayed eye” button to the right of the molecule definition area (NOT the component definition area). Once all the molecules are hidden, click the “eye” button to the right of the component definitions. Then zoom in to see the 4 “sticks” that show the layout of these components at Blender’s origin. It’s helpful to hide the manipulator and move the 3D cursor to see it better.

You should see 4 orange lines radiating out from the origin. Three of these (in the x-y plane) are actual components. The fourth (vertical) line is the rotation reference key that we defined along the z axis. Now we can start experimenting.
Click the “+” button in the component section again to add another component. It will appear below the rotation key, and will have its “Rot Ref” column set to the default of -1. Change that rotation reference to “3” (or use the “key” button) just like the other actual components. Again click the “2D” button to assign location to all of the components, and again click the “eye” button in the components subpanel to show the new arrangement. It should show four “stick” lines in the x-y plane and one rotation reference “stick” along the “z” axis.
Now click the “3D” button to assign 3D locations to the components. You will see non-zero values in every row of the “Loc: z” column. Click the “eye” button to see the arrangement. You should see the points arranged to form a tetrahedron (2 in the x-z plane and 2 in the y-z plane). You’ll also see the vertical rotation reference along the “z” axis.

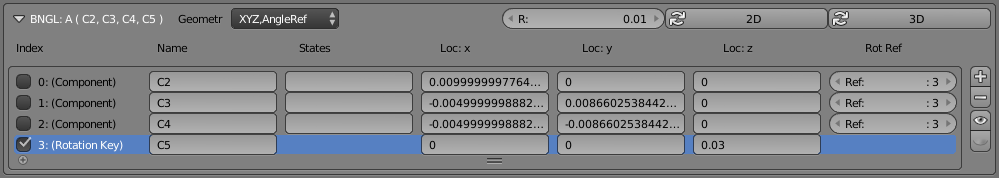
Add another component with the “+” button, and again set its “Rot Ref” column to “3” to use our existing rotation reference. Click the “3D” button, and click the “eye” to see the new distribution. In this case, there will be 3 sticks arranged in the x-y plane, and one in the negative “z” axis, and one in the positive “z” axis. You won’t see the positive “z” component because it’s hidden by the reference. To prove that it’s really there (and provide a nice spiral effect later), change the “Loc x” value of the Rotation Key (item 3) to be 0.01. That will move it slightly off the “z” axis. Then click the “eye” to show the new configuration.

Let’s add 5 more components (click the “+” button 5 times). Change each of the new “Rot Ref” fields to “3” (or use the “key” button) as we did with the other components. Click the “3D” button to assign them values, and then click the “eye” button to see the result. You should see 10 component lines and one rotation reference line.

You can change the radius of these component locations with the “R” field at the top of the box. It’s normally defaulted to 0.01, but you can change it and click the “3D” button to assign the new values, and then click the “eye” button to see the result. Change it back to 0.01 and again click the “3D” button when you’re done. Also hide the “sticks” with the “grayed eye” button.
So far, we’ve just been using the “quick preview” feature of the structured molecule panel. To see a more proper 3D version of our molecule, open the “Molecule Structure Tool” panel. The top row will show an empty “MolDef” field. That’s where you can type in a single molecule name or a complex. Start with just the molecule name of “A”. Then click the “down arrow chevrons” to the right. That will fill out the panel with entries for each component available in your complex. Since this is just a single molecule, you’ll see “Molecule A” in entry “0” followed by all of its components and keys.
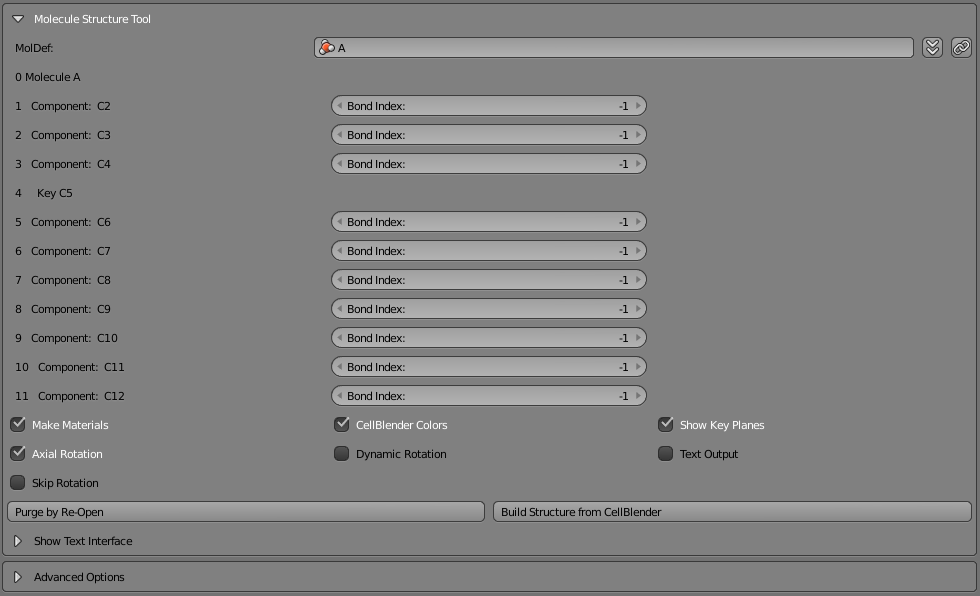
We’ll discuss this panel further when we begin to build complexes. For now, just click the “Build Structure from CellBlender” button at the lower right of the panel. You should see a nice rendering of your 10-component molecule along with the rotation key and semi-transparent rotation key planes between each component and its rotation key. Zoom in and look around to get an understanding of what’s there.
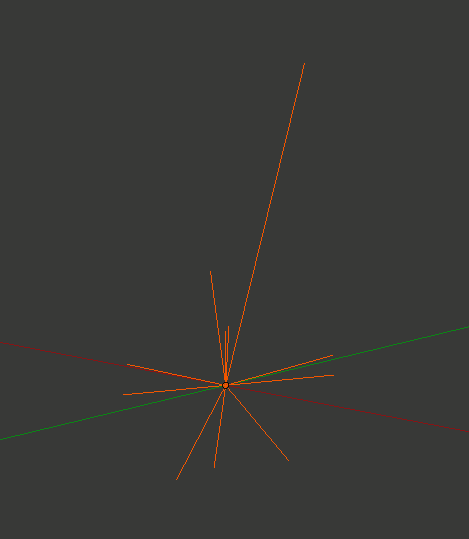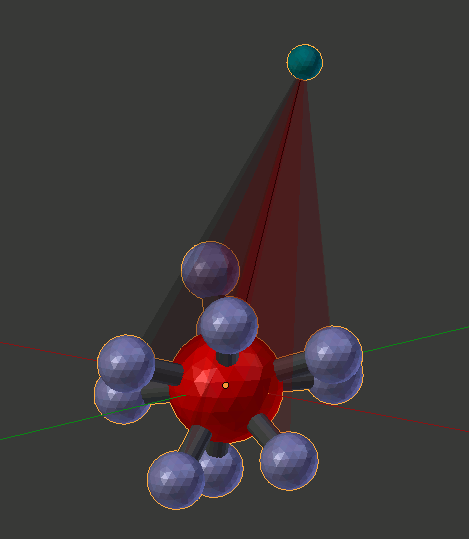
At this time, the Molecule Structure tool generally doesn’t delete what it makes (except in “Dynamic Rotation” mode). This allows you to drag your creations off to the side or hide them and make others. For this tutorial, we’re generally going to delete everything after we’ve made it, so go ahead and use your favorite method to delete everything from the scene (the “a” and “x” keys are a good choice).
Now go back to the “MolDef” entry and replace the single “A” with “A.A”. As before, click the “down chevrons” to fill the panel with the components of this new complex. If you click the “Build Structure from CellBlender” button, you’ll still only see a single molecule. But in reality, there will be two molecules there on top of one another (if you’re familiar with Blender, you can go into edit mode and start disassembling it to convince yourself that there are two of everything in that mesh). To get the molecules to form a structure, we’ll have to create bonds between some of the components.
Start by deleting everything from the scene again. Then go to the Molecule Structure Tool and click the “chain” button at the right of the top row.
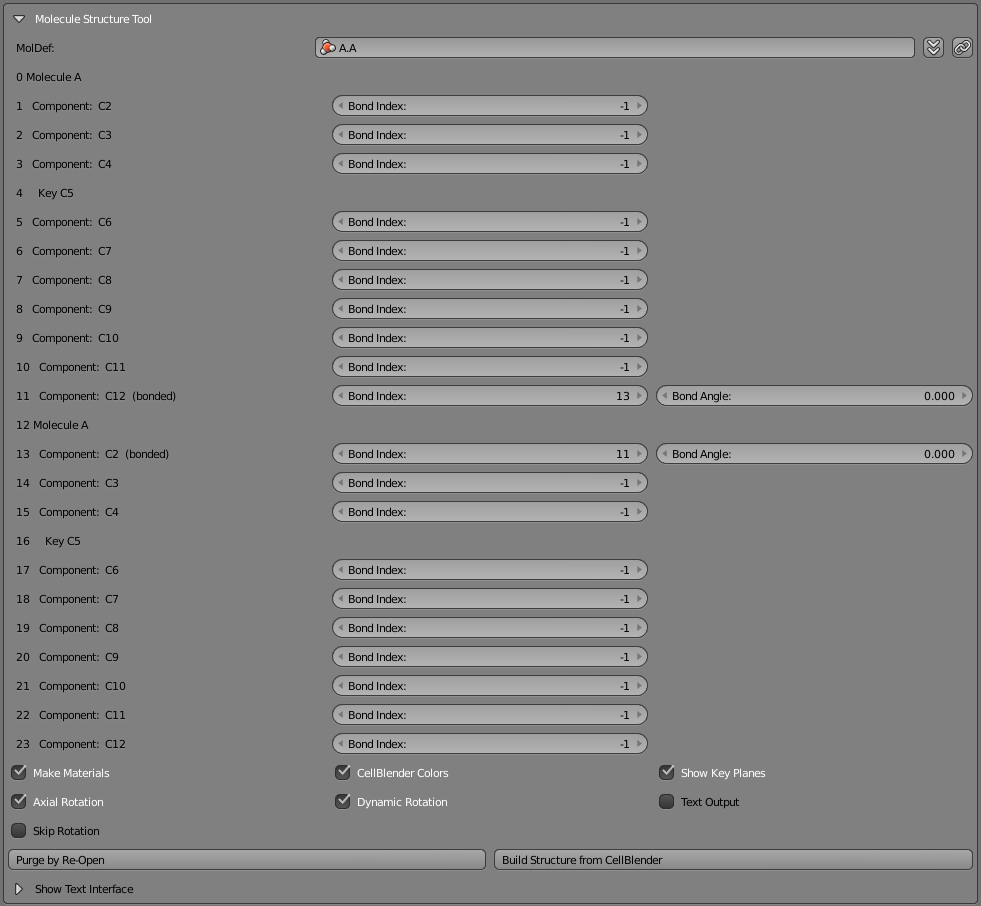
You’ll notice that two “Bond Angle” entries show up attached to the last component of the first molecule and the first component of the second molecule. You’ll also notice that the “Bond Index” values for those two components reference each other while all the other “Bond Index” values are -1. That’s how this tool knows that two components are connected. They reference each other. Now click the “Build Structure from CellBlender” button and you should see two of these new molecules bonded together. If your “A” molecule has 10 components, you’ll find that component index 11 is connected to component index 13 and component index 13 is connected to componenent index 11. Both of these will show a Bond Angle of 0.
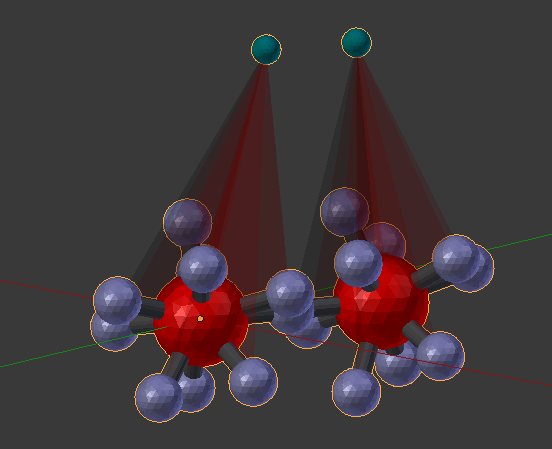
Let’s rotate the second molecule with respect to the first. If you somewhat line up the two molecules in Blender’s 3D view, you’ll notice that their rotation references will also line up (small blue-green spheres). Click the “Dynamic Rotation” check box, and then change the first “Bond Angle” to 0.3.
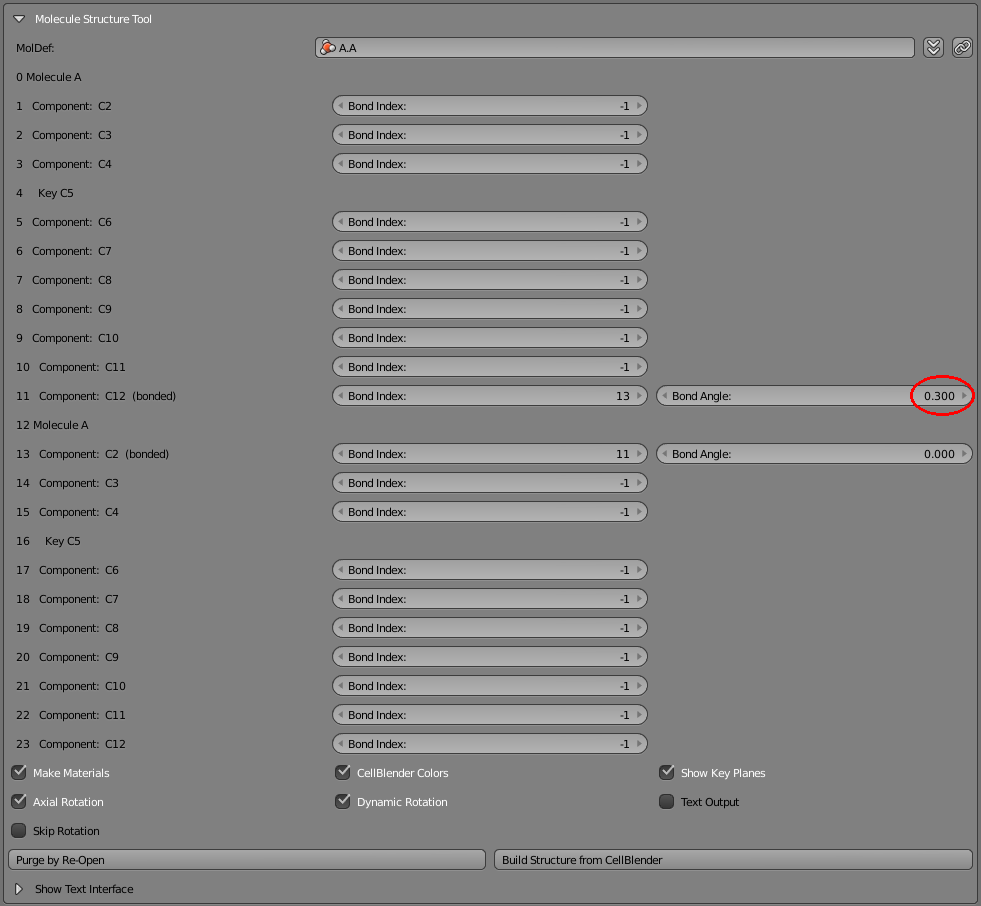
You should notice that the second molecule you added is rotated 0.3 radians. Now try 3.14, and the second molecule will be rotated 180 degrees from the first. You can also click and drag within that same “Bond Angle” entry field to dynamically rotate the second molecule by holding down your mouse button and sliding to the left and right.
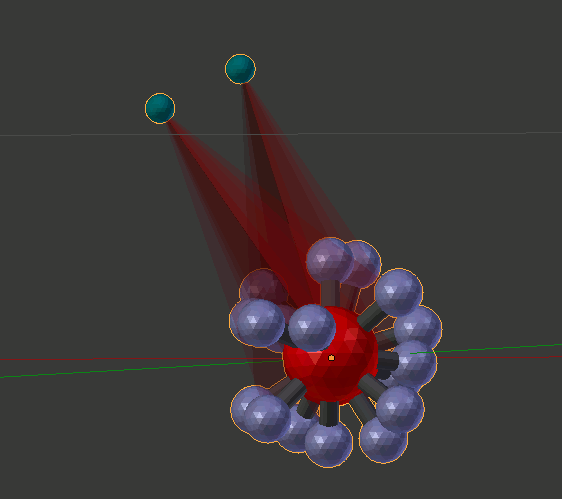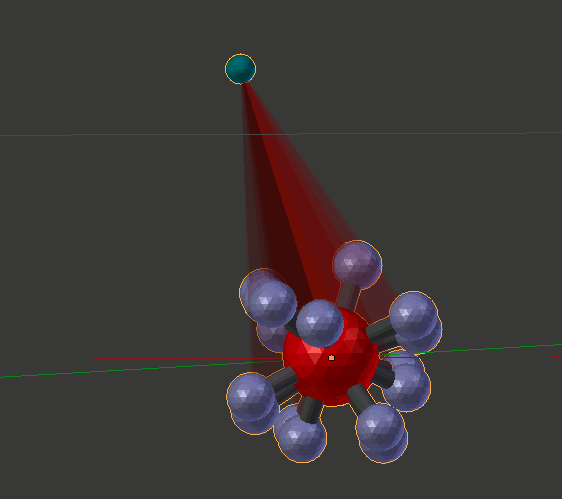
Note that after a short while, the animation might begin to slow down. This is an unresolved problem caused by accumulating lots of copies that are not automatically purged. If the animation slows down too much, just click the “Purge by Re-Open” button in the lower left of the panel. That will speed it up until you again accumulate too many frames of data with lots of dragging.
Now we can begin to make larger complexes. Delete everything in the scene, turn off the “Dynamic Rotation”, and then enter “A.A.A.A.A” (5 A’s) into the MolDef field. Click the “down chevron” to fill out the panel, and then click the “chain” to bond them all end-to-end. Build the actual complex with the “Build Structure from CellBlender” button as before. You should see 5 molecules all strung together. It just so happens that the first and last component of each of these molecules is about 180 degrees apart from each other, so the binding sites tends to create a long slightly curved chain.
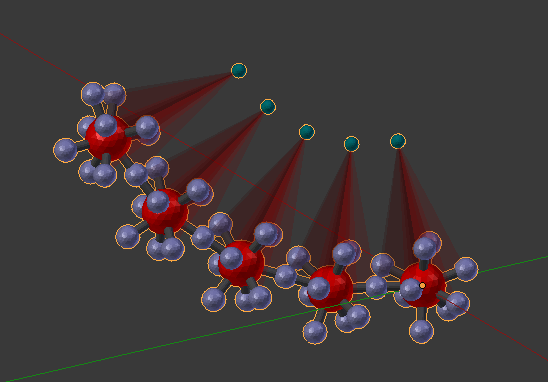
We can edit the structure of this complex by breaking and creating bonds. But this model is a bit too complex for a first model. So let’s start over with a set of simpler molecules. Delete everything in the scene, and go back to the original “Defined Molecules” panel. Let’s peel off some of those compoents from the “A” molecule by clicking the “-” button when the last component is highlighted. Remove all components except 0,1,2,3. Then click the “3D” button to recalculate coordinates for the components. Go back down to the Molecule Structure Tool and repopulate the panel with the “chevron” button, and chain them all together with the “chain” button. Click the “Build Structure from CellBlender” button again, and you’ll find that you’ve got 5 molecules in part of a spiral screw shape.
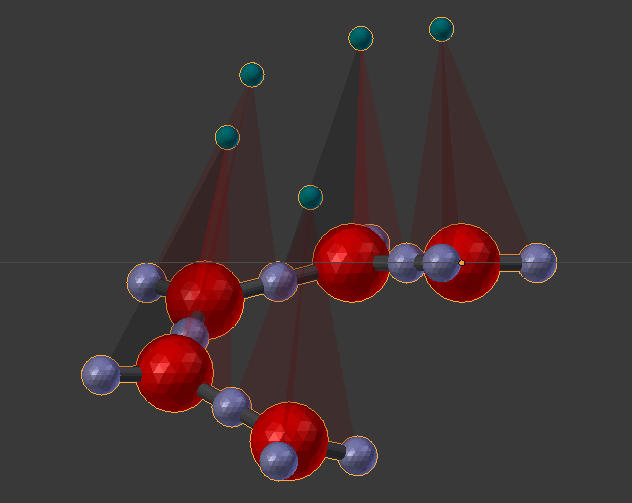
The circular aspect of this shape obviously comes from the 120 degree angles between the components in each of these simpler molecules. The spiral “screw” shape comes from the slight tilt (x=0.01) that we gave to our Rotation Reference Key when we defined the “A” molecule. If we had placed that Rotation Reference Key on the “z” axis, then the partial ring of molecules would be flat.
Let’s add 3 more “A” molecules to our complex to get “A.A.A.A.A.A.A.A” for a total of 8. Remember to delete the current molecule(s) from the 3D view and then repeat the process of clicking the “chevron” to fill out the panel, the “chain” to bind them together, and finally, the “Build Structure from CellBlender” to build the actual complex. As expected, you should see a similar spiral with just a few more parts.
If you continued to add “A” molecules, the structure could grow indefinitely. Here’s an example with 32 “A” molecules:
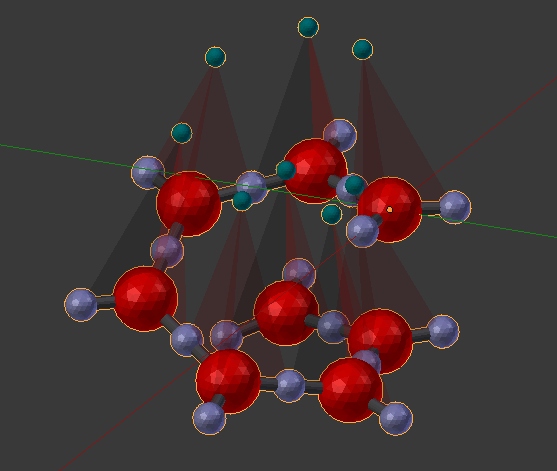
Now we’re going to disconnect part of our 8 molecule chain and re-attach it at a different location. Let’s start by breaking the molecule in the middle. The two halves are joined by the bonds at 18 and 21 which reference each other. Here’s what they look like before breaking:
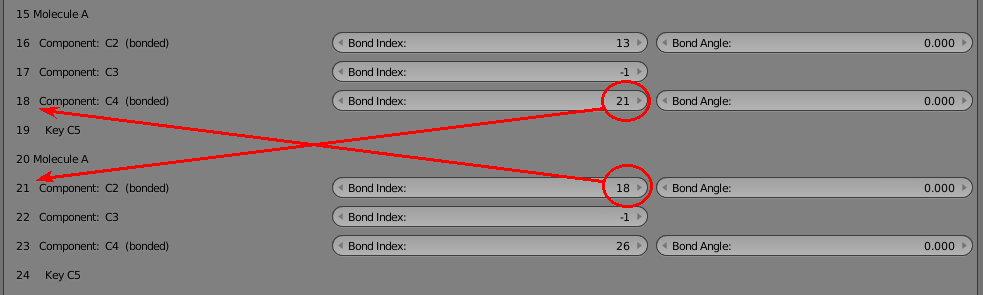
Change them both to -1 to indicate that they are not bound. You’ll notice that a component with an inconsistent bond (referencing another that doesn’t reference back) shows up as red. This is very helpful when manually reconfiguring bonds in this tool.
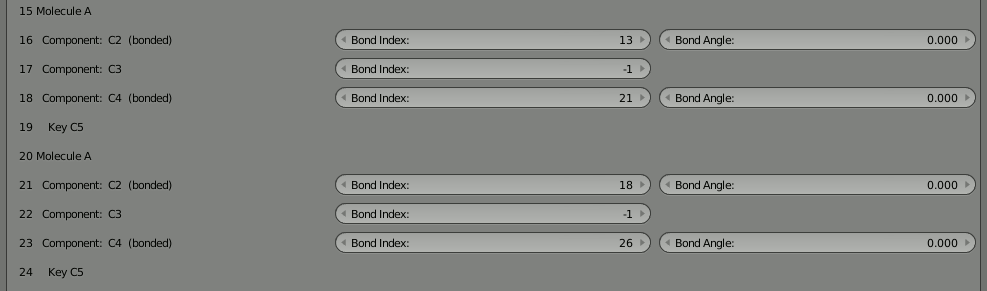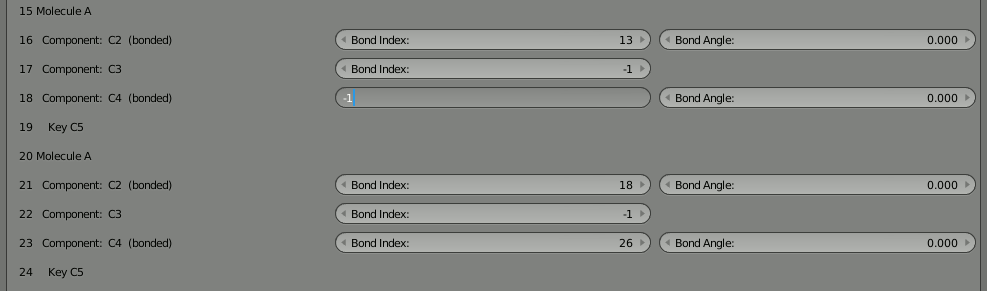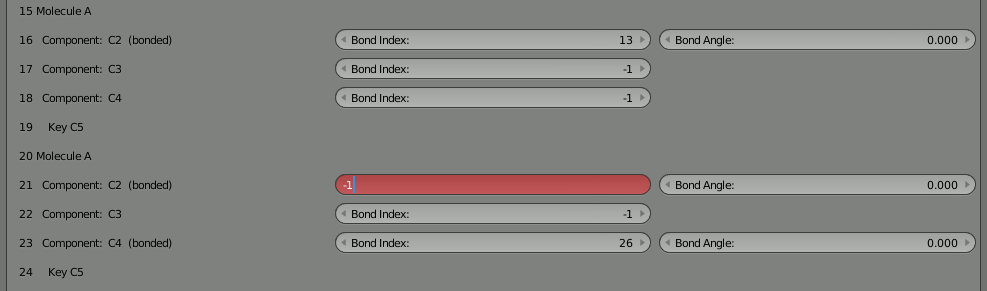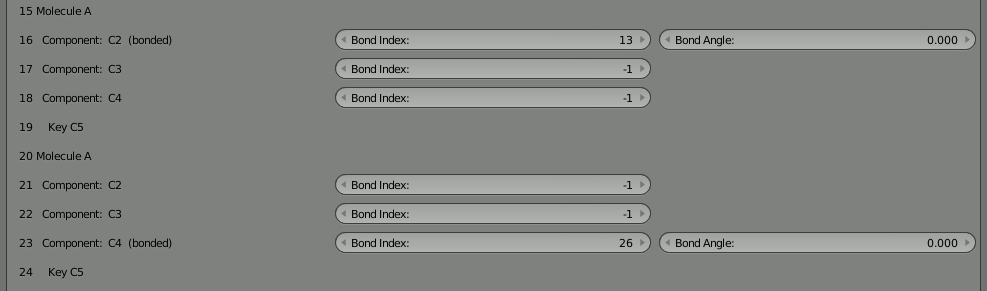
With the bonds broken, you can directly click the “Build Structure from CellBlender” button to show the two parts. Be sure NOT to click the chevron or the chain because that will either break all the bonds or reconnect the full chain. You’ll notice that the two parts only show up as one. As before, if you disassemble this molecule in Blender’s “Edit Mode” you’ll find that there are duplicates of everything because both parts are positioned on top of each other.
To rejoin the two parts, we need to decide which unbound components can be used to rebind the complex. We could pick any unbound components, but let’s choose 12 and 27 since they’re more toward the middle of our 4 molecule segments. So put “12” into the “Bond Index” field for 27, and put “27” in the “Bond Index” field for 12. You’ll notice the red warning letting you know the bond isn’t completed. It should go away when the bond is correct.
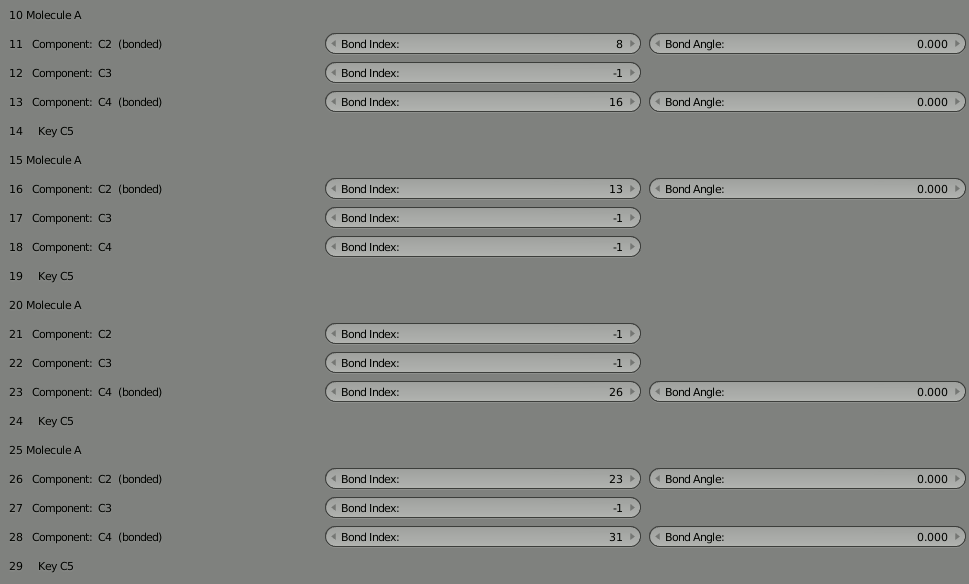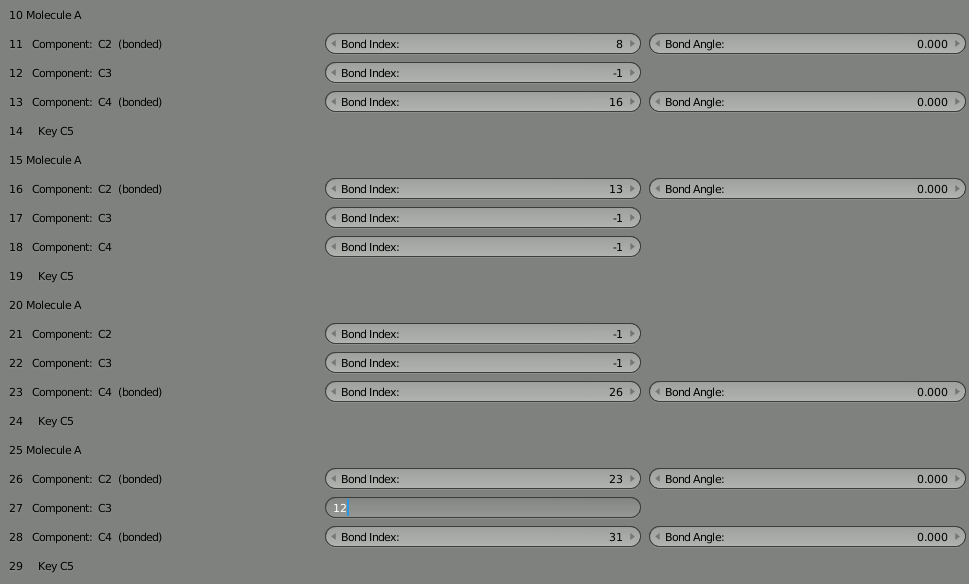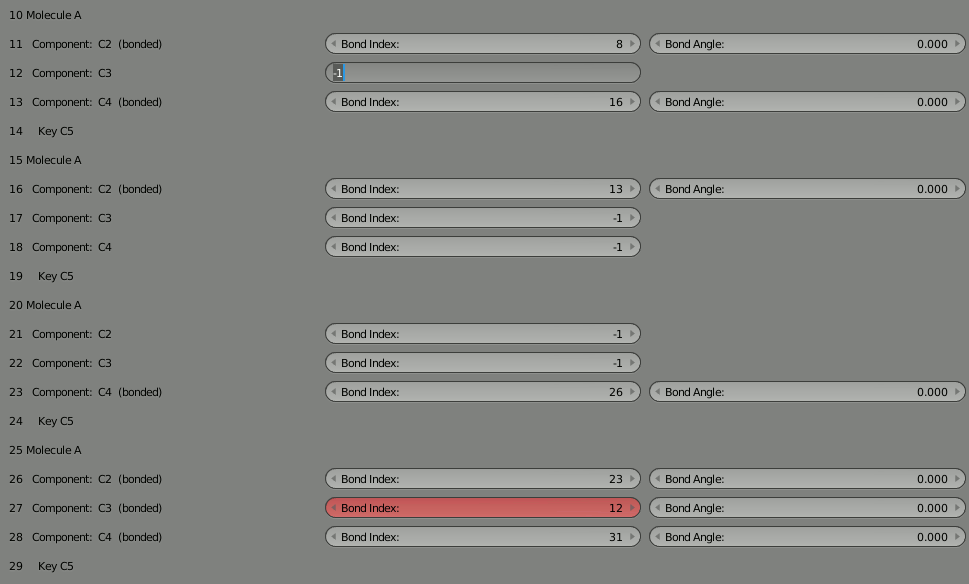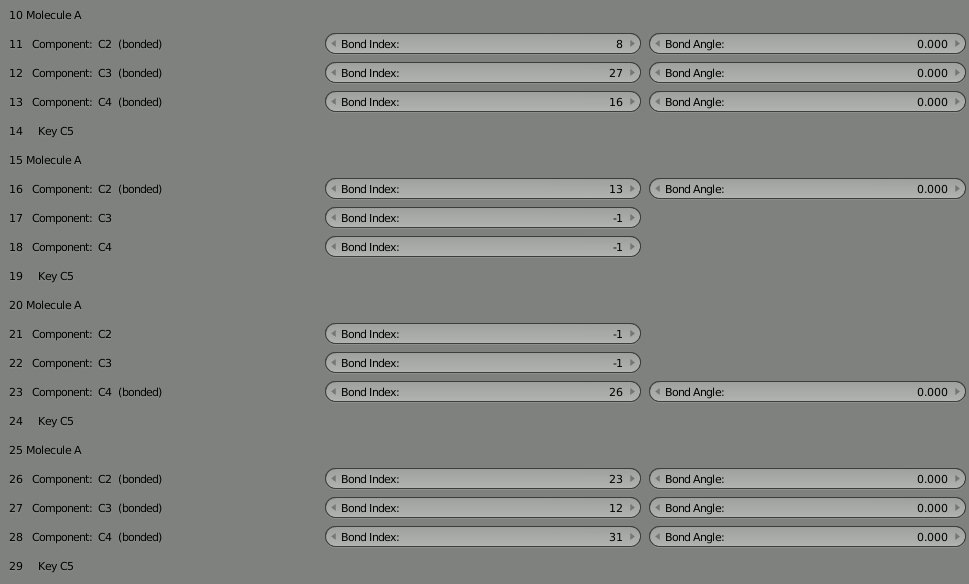
As usual, delete the molecule in the 3D view and then click “Build Structure from CellBlender” to see the result. Take some time to look at the complex. Notice how the two parts are now joined.
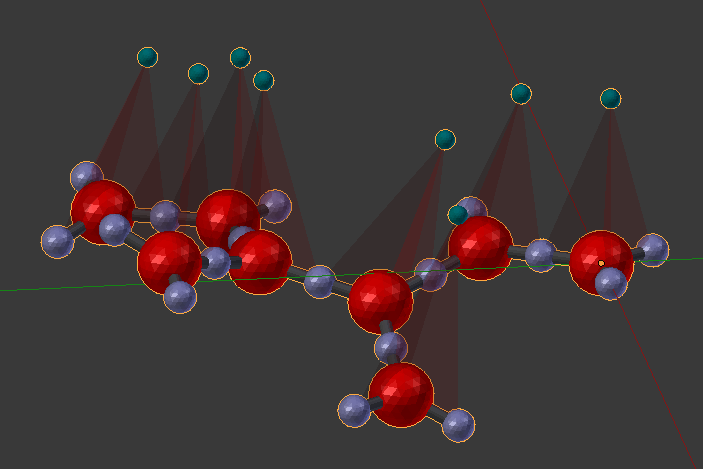
Also notice the Rotation Key Planes at the joint. You should see that the two planes are aligned with each other (a rotation angle of 0). Enable “Dynamic Rotation” with the check box, and begin to rotate the angle on component 27. Rotate it to a value of about 2.0 which will make the two sections somewhat perpendicular to each other.
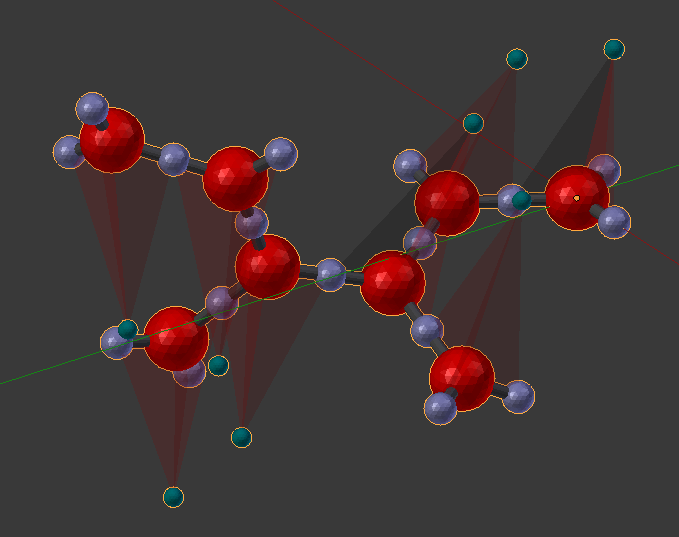
When you’re done, disable the “Dynamic Rotation” check box, and click the “Purge by Re-Open” button to remove any stale data. At this point, the Rotation Angle “Key” planes are just in the way, so disable that check box (“Show Key Planes”) as well. Then delete the object and build it again with the same “Build” button we’ve been using (“Build Structure from CellBlender”). You should see a nice clean version of your complex without the alignment planes in the way.
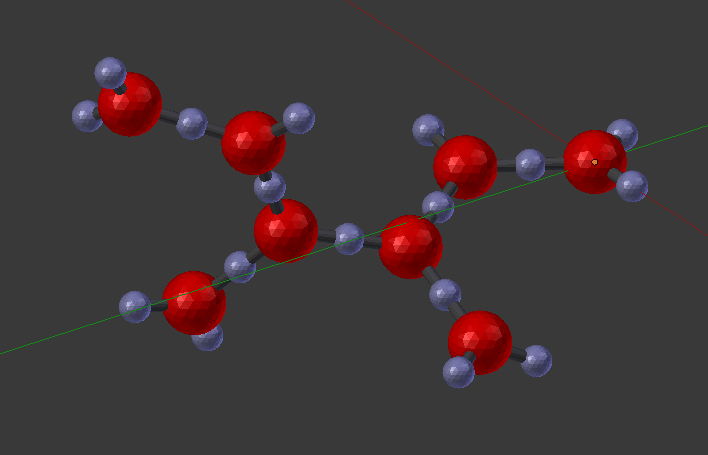
There are a number of other tools available in this panel. One easy one to try is disabling the 3D rotation. For this example, don’t delete the previous complex. Instead, click on it and drag it away from the center up the “z” axis. This is easily done with the “g” hot key followed by the “z” hot key. That will constrain your mouse to drag it only along the “z” axis. Drag it up about as high as it is wide. Then click the “Axial Rotation” check box to turn it off. Then build the molecule again (“Build Structure from CellBlender”). This will build the same molecule, but without axial rotation, all of the bonds will be flat. That’s a side effect of the fact that our 3 component molecules are already flat (in a plane). With planar molecules and no axial rotations, the result will also be in a plane.
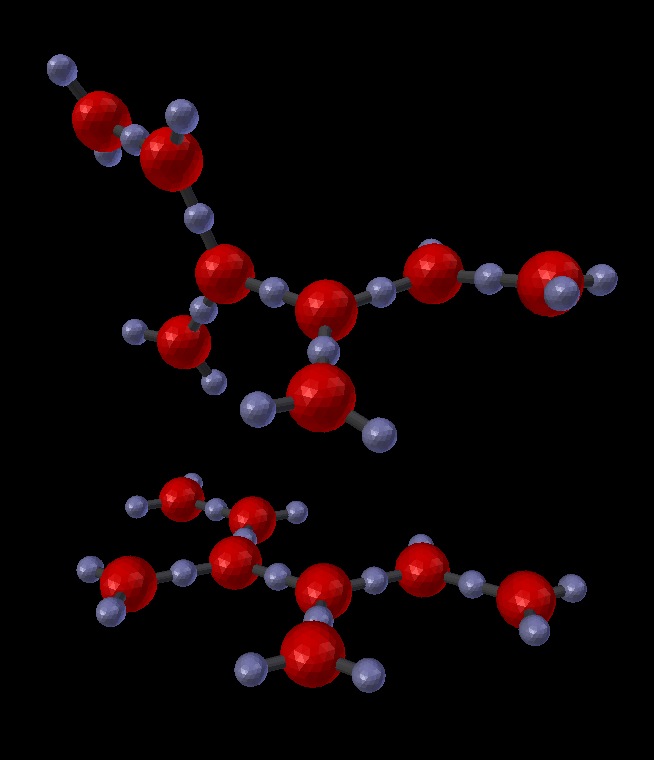
Another handy option is the “Average Coincident” switch. The current binding model specifies that when two components bind, they occupy the same point in space. In other words, component locations are the joining points for molecules. This model works naturally when there are no loops, and all molecules and components end up wherever their geometry dictates. However, when there are loops, there is normally no guarantee that the closing points of a loop are actually coincident in space. For example, if a molecule were designed as a square (four molecules each binding to each other with components at 90 degrees), then the geometry would dictate that the closing points would be coincident. But if the angles were specified (incorrectly) as 91 degrees rather than 90, then the “closing” components would not actually join. This could be considered as either a fundamental error in the model or as a minor round off error. While that decision is up to the designer, the current tool does provide a simple method to “fix” any such “round off” errors using the “Average Coincident” option. The “Average Coincident” option performs one final step after building the molecule. It sets the location of each partner of a binding pair to be the average location of the two binding partners. For small rounding errors, this simply brings the points into exact coincidence. However, for gross errors (such as forming an equilateral triangle with right angles), it will drastically change the geometry. The decision to use this feature (or not) is up to the model’s designer. This effect can be seen in the following image where the geometry was intentionally distorted to misalign the closing bond:
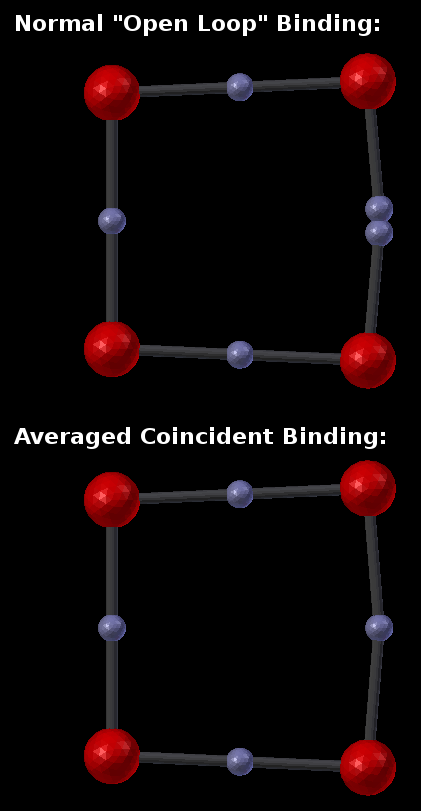
As seen in this example, the “Average Coincident” option forces the two components to be at the same point in space. Note that this does not correct any other deficiencies of the model (such as the misalignment across the bond). Note also that this is a purely spatial effect. The actual binding of a molecule is specified in the structure of its BNGL definition. That definition is inherently non-spatial. So even though the components may not “close” properly, if the componets are bonded, then they will behave as such.
14.5.1.2.1. Conclusion¶
This tutorial has covered the basics of creating spatially structured molecules and complexes in CellBlender. Using these tools, almost any shape can be approximated. Note that we use the word “approximated” because the molecules built in CellBlender are only intended to provide approximate structure. But this approximate structure is useful for simulating many of the spatial aspects of such molecules within stochastic simulators like MCell. It’s also important to note that while rudimentary complexes may be built by hand (as in this tutorial), the real power behind spatially structured molecules arises from rule based simulations which can build these emergent structures automatically.

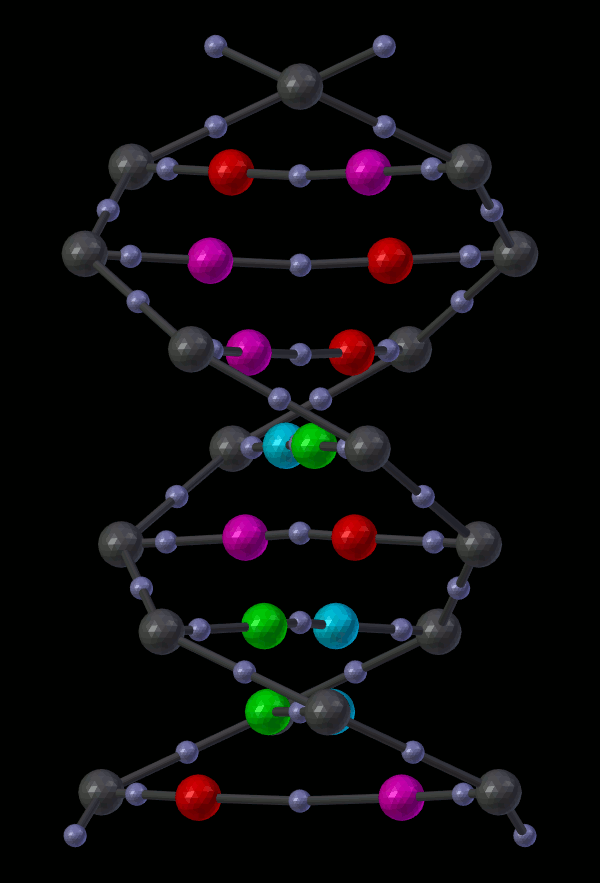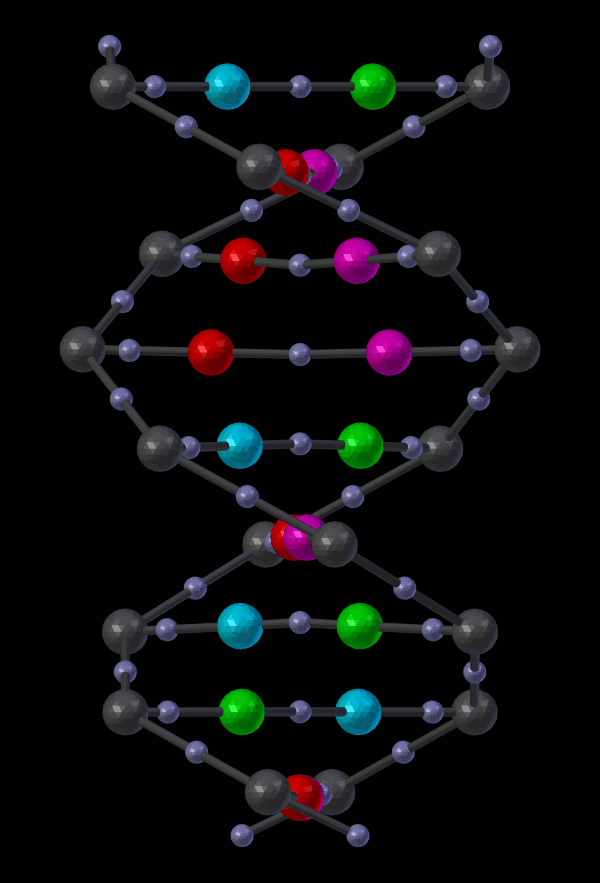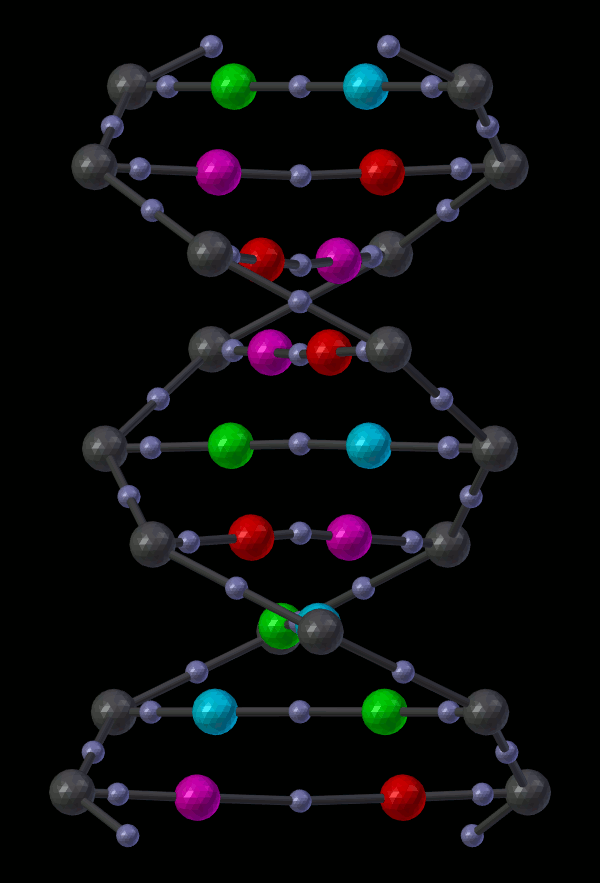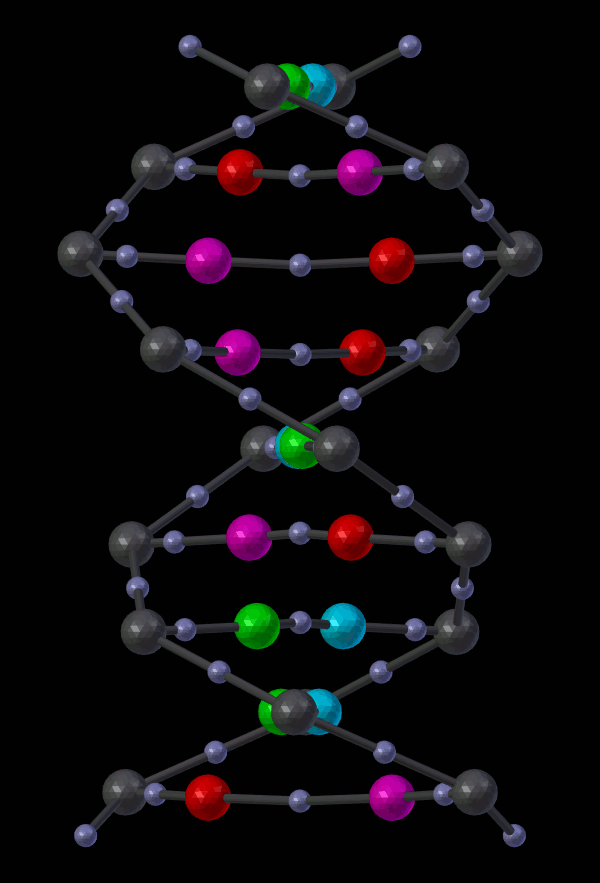
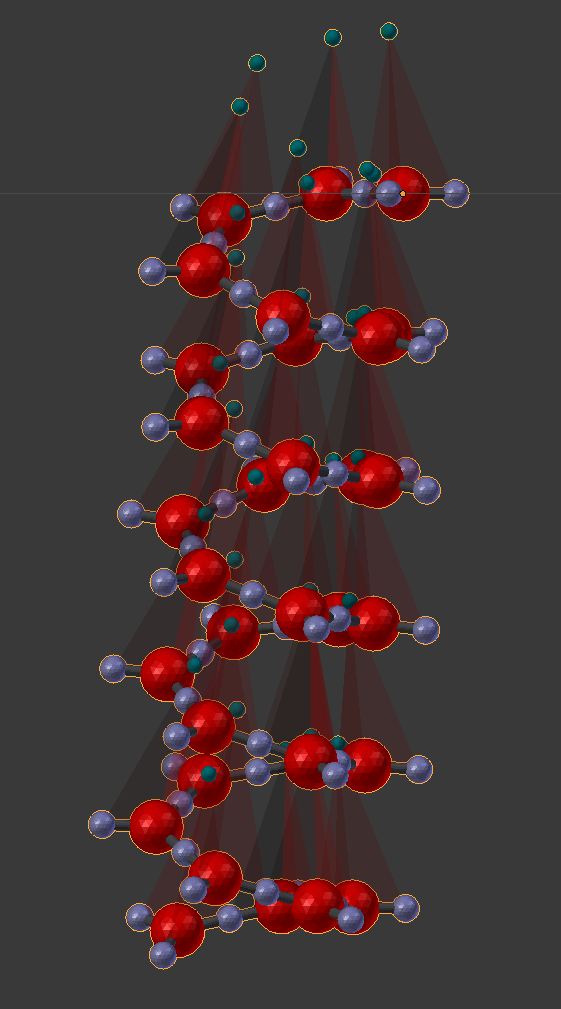
14.5.1.2.2. Appendix - Source code for Double Helix model¶
The Double Helix model shown above was constructed from a Python script that modifies CellBlender’s internal properties directly. This is not generally a good practice, and the preferred scripting method is to modify a CellBlender data model. However, at this stage of development, the direct CellBlender method was easier and is presented here.
To use this code, open a blender “Text” window and create a new file. Name the file “Double_Helix.py”, and copy the following code into it. Be sure that all of the requirements are satisfied (BioNetGen Language Mode on, and A,C,G,T,S molecules as specified in the comments). Be sure to check everything carefully. Any mistakes are likely to cause difficulty. Then click the “Run Script” button in the “Text” window. That should populate the “Molecule Structure Tool” with all the molecules. Once that’s done, you can choose whether you want to see key planes or not and whether you want to average coincident points or not. Then click the “Build Structure from CellBlender” button in the “Molecule Structure Tool” panel. It may take a few seconds to do all the calculations.
Eventually, the double helix molecule should appear in the 3D view window similar to what’s shown above. It will be fairly small, so you may have to zoom in to the origin to see it. The molecule colors may be different, but you can change those using the normal Blender molecule material properties or in CellBlender’s Molecule / Display Options panel. For the animation shown here, “G” was assigned Green, and “C” was assigned Cyan (as easy mnemonics). The “A” was assigned red, and the “T” was assigned magenta.
# Double Helix Construction Script
# This script works directly with CellBlender's internal data
# structures to build a double helix molecule similar to DNA.
# This script relies on the following CellBlender Definitions:
#
# Parameter "r" = 0.0175
#
# Molecules and Components:
#
# Molecules "A", "C", "G", and "T" are all defined with components:
# Ac1: x=r y=0 z=0 Ref=2
# Ac2: x=-r y=0 z=0 Ref=2
# Ak: x=0 y=0 z=0.008 (Reference Key)
#
# Cc1: x=r y=0 z=0 Ref=2
# Cc2: x=-r y=0 z=0 Ref=2
# Ck: x=0 y=0 z=0.008 (Reference Key)
#
# Gc1: x=r y=0 z=0 Ref=2
# Gc2: x=-r y=0 z=0 Ref=2
# Gk: x=0 y=0 z=0.008 (Reference Key)
#
# Tc1: x=r y=0 z=0 Ref=2
# Tc2: x=-r y=0 z=0 Ref=2
# Tk: x=0 y=0 z=0.008 (Reference Key)
#
# Molecule "S" is defined with components:
# Sc1: x=-0.02 y=0.0084 z=0 Ref=3
# Sc2: x=0 y=0.01 z=0 Ref=3
# Sc3: x=0.02 y=0.0084 z=0 Ref=3
# Sk: x=-0.001 y=0 z=0.008 (Reference Key)
#
# This script can be run with the "Run Script" button in the editor.
# It will overwrite the values in the "Molecule Structure Tool".
# After running this script, press the "Build Structure from CellBlender" button.
#
# The half strand of the sequence can be changed arbitrarily:
half_strand = 'CATTGACGA'
# This angle is specified in radians
end_cap_angle = 0.3
# This is the number of segments between base pairs (use 1 for now)
back_bone_segments_per_base_pair = 1
# These are used to look up partners (don't normally change)
nucleotide_partner = {'G':'C', 'C':'G', 'A':'T', 'T':'A'}
import bpy
# Import and assign the Blender and CellBlender data
mcell = bpy.context.scene.mcell
molmaker = mcell.molmaker
molcomp_list = mcell.molmaker.molcomp_items
# Create a "dotted" specification that includes both base pairs for the molecule definition
molmaker.molecule_definition = '.'.join ( [ c + '.' + nucleotide_partner[c] + '.S.S' for c in half_strand ] )
# Clear out the old molcomp_list
while(len(molcomp_list) > 0):
molcomp_list.remove(0)
# Build the Double Helix base pairs from the half strand data
cur_mol_index = 0
# Start with just the first pair (treating it as the base)
for i in range(1):
print ( "Building first base pair for " + half_strand[0] )
# Make a pair of Nucleotides
for n in range(2):
nucleotide_name = half_strand[i]
if n > 0:
nucleotide_name = nucleotide_partner[nucleotide_name]
# Make this Double Helix Nucleotide
new_mol = molcomp_list.add()
new_mol.name = nucleotide_name
new_mol.field_type = 'm'
new_mol.alert_string = ''
new_mol.peer_list = ''
new_mol.peer_list = str(cur_mol_index+1)+','+str(cur_mol_index+2)+','+str(cur_mol_index+3)
# Add Binding Components
for j in range(2):
new_comp = molcomp_list.add()
new_comp.name = nucleotide_name + 'c' + str(j+1)
new_comp.field_type = 'c'
new_comp.alert_string = ''
new_comp.peer_list = str(cur_mol_index)
# Add an Alignment Key
new_key = molcomp_list.add()
new_key.name = nucleotide_name + 'k'
new_key.field_type = 'k'
new_key.alert_string = ''
new_key.peer_list = str(cur_mol_index)
cur_mol_index += 1 + 2 + 1
# Join the two together
molcomp_list[cur_mol_index-3].bond_index = cur_mol_index-6
molcomp_list[cur_mol_index-6].bond_index = cur_mol_index-3
# Make the first end cap
new_mol = molcomp_list.add()
new_mol.name = 'S'
new_mol.field_type = 'm'
new_mol.alert_string = ''
new_mol.peer_list = ''
new_mol.peer_list = str(cur_mol_index+1)+','+str(cur_mol_index+2)+','+str(cur_mol_index+3)+','+str(cur_mol_index+4)
# Add Binding Components
for j in range(3):
new_comp = molcomp_list.add()
new_comp.name = 'Sc' + str(j+1)
new_comp.field_type = 'c'
new_comp.alert_string = ''
new_comp.peer_list = str(cur_mol_index)
# Add an Alignment Key
new_key = molcomp_list.add()
new_key.name = 'Sk'
new_key.field_type = 'k'
new_key.alert_string = ''
new_key.peer_list = str(cur_mol_index)
cur_mol_index += 1 + 3 + 1
# Make the second end cap
new_mol = molcomp_list.add()
new_mol.name = 'S'
new_mol.field_type = 'm'
new_mol.alert_string = ''
new_mol.peer_list = ''
new_mol.peer_list = str(cur_mol_index+1)+','+str(cur_mol_index+2)+','+str(cur_mol_index+3)+','+str(cur_mol_index+4)
# Add Binding Components
for j in range(3):
new_comp = molcomp_list.add()
new_comp.name = 'Sc' + str(j+1)
new_comp.field_type = 'c'
new_comp.alert_string = ''
new_comp.peer_list = str(cur_mol_index)
# Add an Alignment Key
new_key = molcomp_list.add()
new_key.name = 'Sk'
new_key.field_type = 'k'
new_key.alert_string = ''
new_key.peer_list = str(cur_mol_index)
cur_mol_index += 1 + 3 + 1
# Join the first end cap to first nucleotide
molcomp_list[cur_mol_index-17].bond_index = cur_mol_index-8
molcomp_list[cur_mol_index-8].bond_index = cur_mol_index-17
molcomp_list[cur_mol_index-8].angle = end_cap_angle
# Join the second end cap to second nucleotide
molcomp_list[cur_mol_index-3].bond_index = cur_mol_index-12
molcomp_list[cur_mol_index-12].bond_index = cur_mol_index-3
molcomp_list[cur_mol_index-12].angle = end_cap_angle
# Now build the rest as separate strands (not joined)
next_base_pair_index = 1
last_backbone_1_index = 11
last_backbone_2_index = 16
i = 0
while next_base_pair_index < len(half_strand):
print ( "Building next base pair for " + half_strand[next_base_pair_index] )
# Add to the backbone
for back_bone in range(back_bone_segments_per_base_pair):
# Make the first end cap
new_mol = molcomp_list.add()
new_mol.name = 'S'
new_mol.field_type = 'm'
new_mol.alert_string = ''
new_mol.peer_list = ''
new_mol.peer_list = str(cur_mol_index+1)+','+str(cur_mol_index+2)+','+str(cur_mol_index+3)+','+str(cur_mol_index+4)
# Add Binding Components
for j in range(3):
new_comp = molcomp_list.add()
new_comp.name = 'Sc' + str(j+1)
new_comp.field_type = 'c'
new_comp.alert_string = ''
new_comp.peer_list = str(cur_mol_index)
# Add an Alignment Key
new_key = molcomp_list.add()
new_key.name = 'Sk'
new_key.field_type = 'k'
new_key.alert_string = ''
new_key.peer_list = str(cur_mol_index)
cur_mol_index += 1 + 3 + 1
# Make the second end cap
new_mol = molcomp_list.add()
new_mol.name = 'S'
new_mol.field_type = 'm'
new_mol.alert_string = ''
new_mol.peer_list = ''
new_mol.peer_list = str(cur_mol_index+1)+','+str(cur_mol_index+2)+','+str(cur_mol_index+3)+','+str(cur_mol_index+4)
# Add Binding Components
for j in range(3):
new_comp = molcomp_list.add()
new_comp.name = 'Sc' + str(j+1)
new_comp.field_type = 'c'
new_comp.alert_string = ''
new_comp.peer_list = str(cur_mol_index)
# Add an Alignment Key
new_key = molcomp_list.add()
new_key.name = 'Sk'
new_key.field_type = 'k'
new_key.alert_string = ''
new_key.peer_list = str(cur_mol_index)
cur_mol_index += 1 + 3 + 1
print ( "Ready to join backbone with cur_mol_index = " + str(cur_mol_index) )
print ( " Last BB1 = " + str(last_backbone_1_index) )
print ( " Last BB2 = " + str(last_backbone_2_index) )
print ( " Should connect " + str(last_backbone_1_index) + " and " + str(cur_mol_index-9) )
print ( " Should connect " + str(last_backbone_2_index) + " and " + str(cur_mol_index-4) )
# Join the first end cap to the previous first nucleotide
molcomp_list[last_backbone_1_index].bond_index = cur_mol_index-9
molcomp_list[cur_mol_index-9].bond_index = last_backbone_1_index
molcomp_list[cur_mol_index-9].angle = end_cap_angle
last_backbone_1_index = cur_mol_index-7
# Join the second end cap to second nucleotide
molcomp_list[cur_mol_index-4].bond_index = last_backbone_2_index
molcomp_list[last_backbone_2_index].bond_index = cur_mol_index-4
molcomp_list[cur_mol_index-4].angle = end_cap_angle
last_backbone_2_index = cur_mol_index-2
# Add another pair of Nucleotides
for n in range(2):
nucleotide_name = half_strand[next_base_pair_index]
if n > 0:
nucleotide_name = nucleotide_partner[nucleotide_name]
# Make this Double Helix Nucleotide
new_mol = molcomp_list.add()
new_mol.name = nucleotide_name
new_mol.field_type = 'm'
new_mol.alert_string = ''
new_mol.peer_list = ''
new_mol.peer_list = str(cur_mol_index+1)+','+str(cur_mol_index+2)+','+str(cur_mol_index+3)
# Add Binding Components
for j in range(2):
new_comp = molcomp_list.add()
new_comp.name = nucleotide_name + 'c' + str(j+1)
new_comp.field_type = 'c'
new_comp.alert_string = ''
new_comp.peer_list = str(cur_mol_index)
# Add an Alignment Key
new_key = molcomp_list.add()
new_key.name = nucleotide_name + 'k'
new_key.field_type = 'k'
new_key.alert_string = ''
new_key.peer_list = str(cur_mol_index)
cur_mol_index += 1 + 2 + 1
print ( "Ready to join base pair with cur_mol_index = " + str(cur_mol_index) )
# Join the two to the backbone
molcomp_list[cur_mol_index-7].bond_index = cur_mol_index-16
molcomp_list[cur_mol_index-16].bond_index = cur_mol_index-7
molcomp_list[cur_mol_index-3].bond_index = cur_mol_index-11
molcomp_list[cur_mol_index-11].bond_index = cur_mol_index-3
# The base pairs could be joined together here, but then their
# bonds would be evaluated earlier than the bonds of the outer
# structure. Since that outer structure is intended to dominate,
# these inner bonds will be added last (below).
# The "Average Coincident" switch will force these to be
# coincident anyway regardless of any misalignment. That
# will give a better overall result than binding them here.
next_base_pair_index += 1
i += 1
# At this point, all of the locations should be determined.
# However, the cross-linking between the base pairs (across
# the center of the molecule) has not been done yet.
#
# This last step searches for base pairs (2 components + 1 key)
# and links them according to a known pattern of +4 and -4 from
# the bound end. This pattern was found just by examining the
# structure built up to this point, so any changes to the building
# up to this point should prompt a re-examination of the following
# code.
# Join the base pairs across the middle by looking for broken bonds
first_broken = True
for i in range ( len(molcomp_list) ):
m = molcomp_list[i]
if m.field_type == 'm':
pl = [ int(p) for p in m.peer_list.split(',') ]
print ( "Peer list = " + str(pl) )
if len(pl) == 3:
# This is a base pair molecule with 2 components and a key
# This is potentially one of the half-linked base pairs
if molcomp_list[i+2].bond_index < 0:
# The negative bond index means it's unbound, so bind it
broken_index = i+2
print ( "Found a broken bond for index " + str(broken_index) )
if first_broken:
print ( " First broken: add 4" )
molcomp_list[broken_index].bond_index = broken_index + 4
first_broken = False
else:
print ( " Second broken: add -4" )
molcomp_list[broken_index].bond_index = broken_index - 4
first_broken = True