14.1.4.2. Data Model Scripting Tutorial¶
This tutorial guides you through building a simple script to create a grid of molecules with different diffusion constants and released at different locations in the x-y plane. This tutorial assumes that you are already familiar with building a model using CellBlender, and that you have some (minimal) Python programming experience. It also assumes some familiarity with using Blender (resizing window panels, creating new window panels, and using the Blender text editor).
Note
The scripting interface is relatively new and subject to change.
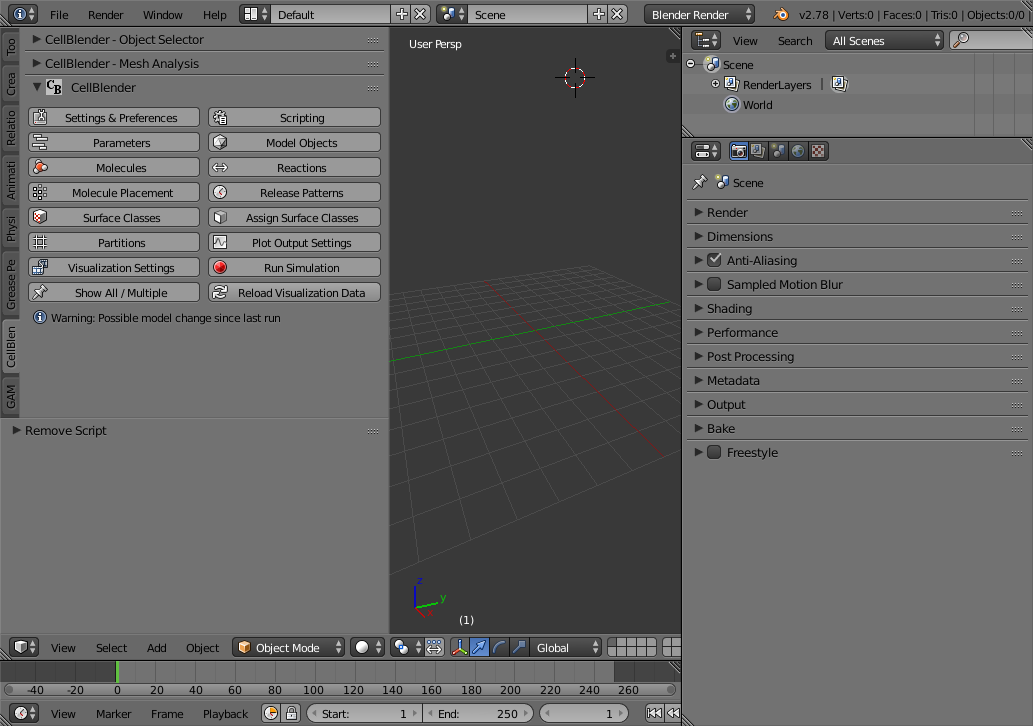
You should start out with a newly initialized CellBlender model with nothing defined (no molecules, reactions, objects, etc). Your screen should look something like this:
The first thing you should do is reconfigure the Blender windows to give yourself a good sized text editor. In this example, the normal “outliner” in the upper right corner has been resized and changed to a text editor containing a new file named “mols.py” (use the “+” button to add the new file, then rename it to “mols.py” as shown below):
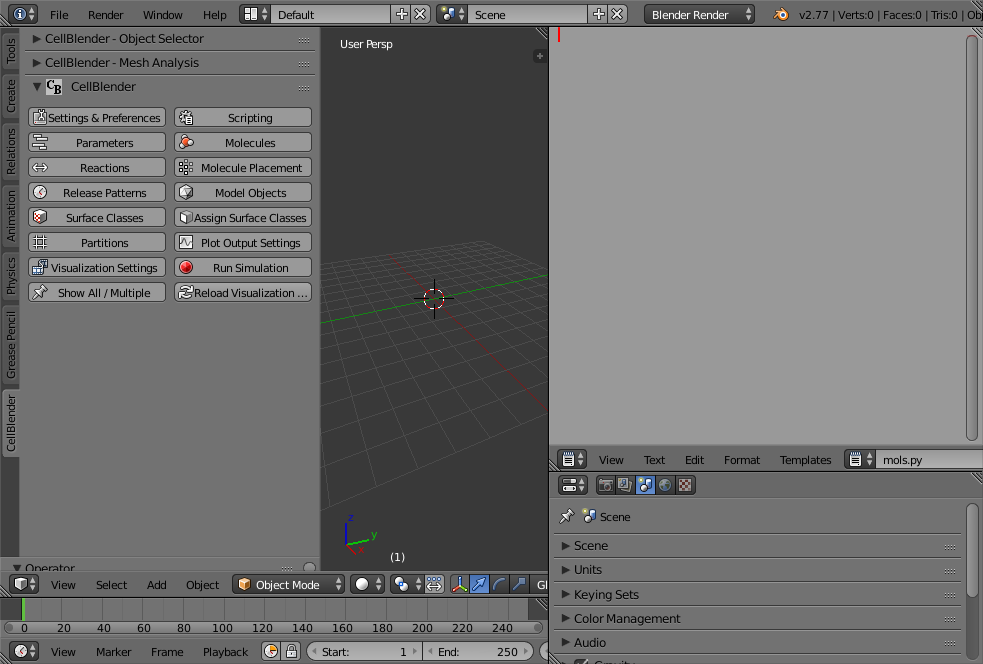
It’s a good idea to name your scripting files to end with “.py” so they can be found in the scripting window for running.
Now enter the following text (discussed below) into that text editing window:
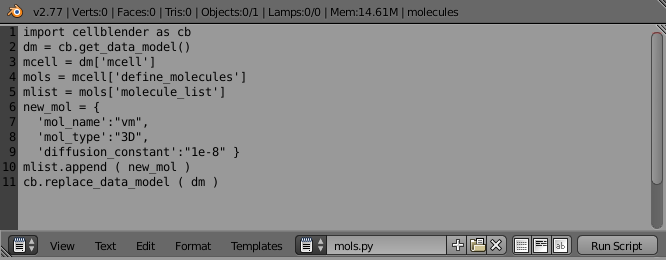
import cellblender as cb
dm = cb.get_data_model()
mcell = dm['mcell']
mols = mcell['define_molecules']
mlist = mols['molecule_list']
new_mol = {
'mol_name':"vm",
'mol_type':"3D",
'diffusion_constant':"1e-8" }
mlist.append ( new_mol )
cb.replace_data_model ( dm )
It should look something like this:
The first line in that script imports “cellblender” using the local alias “cb”. The cellblender module contains 4 handy functions for data model scripting. By importing “cellblender” as “cb”, these 4 functions will be available as “cb._______” as shown below:
Handy Functions for Data Model Scripting:
Function
Description
cb.get_data_model(geometry=False)
Returns a copy of the current CellBlender Data Model
cb.replace_data_model(dm,geometry=False)
Replaces the current CellBlender Data Model with dm
cb.cd_to_project()
Changes process directory path to project location (returns previous directory path)
cb.cd_to_location(loc)
Changes process directory path to location passed in by “loc”
The second line of the script uses the “get_data_model” function to get a copy of the current data model from CellBlender. This will be a complete representation of the various data fields that define the current CellBlender model. You can print the data model to the console with print(str(dm)) if you want to see all of its fields (more on this later). For this first example, we won’t use any of the information in the existing data model, but we will add to it.
The 3rd, 4th, and 5th lines assign convenience variables (“mcell”, “mols”, and “mlist”) to various subparts of the data model. This isn’t necessary, but makes the code easier to read.
The 6th through 9th lines define the new molecule as a dictionary with keys for “mol_name”, “mol_type”, and “diffusion_constant”. At this point, you simply have to know that these are the proper key strings, but later, we’ll see how you can obtain them from an existing data model.
The 10th line simply appends the newly created molecule dictionary to the molecule list from line 5. Finally, the last line replaces the CellBlender data model with the modified version named “dm”. This will repopulate all of the Blender/CellBlender properties throughout the CellBlender user interface.
Note that this entire program could reasonably be written in 5 lines like this:
import cellblender as cb
dm = cb.get_data_model()
new_mol = { 'mol_name':"vm", 'mol_type':"3D", 'diffusion_constant':"1e-8" }
dm['mcell']['define_molecules']['molecule_list'].append ( new_mol )
cb.replace_data_model ( dm )
The choice of creating convenience variables (as in the earlier example) or not is mostly a matter of programming style.
You’ll notice that there’s a “Run Script” button in the header below the text editing window. This can be used to run a data model script, but we will use the “Run Script” button in the scripting panel. Start by opening the Scripting Panel as shown here:
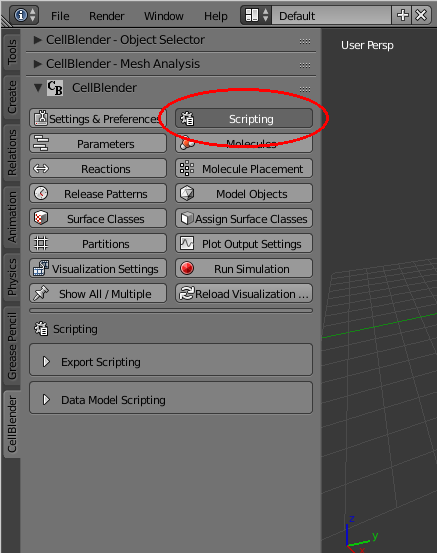
Then expand (open) the “Data Model Scripting” and “Run Script” subpanels, and click on the “refresh” button (to reload the File selector):
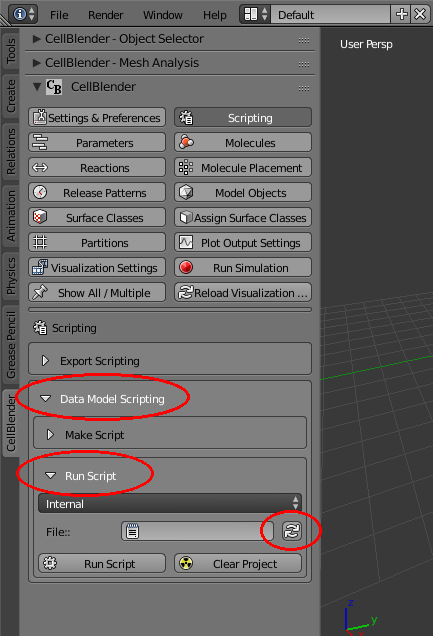
Then click on the “File” selector and choose the “mols.py” file:
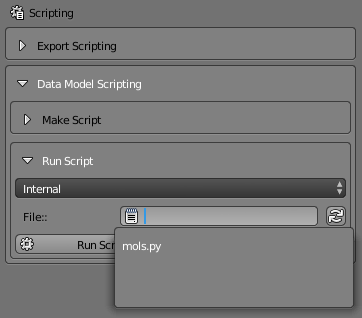
Before running the script, let’s show both the molecules panel AND the scripting panel at the same time. You can do this by clicking the “Show All/Multiple” push pin button and then selecting both the Scripting and Molecules buttons:
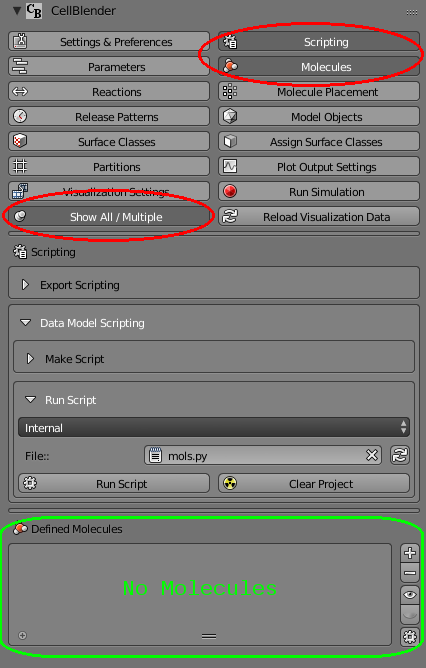
You’ll notice that the “Defined Molecules” section is empty (No Molecules) before you run the script. Then when you click the “Run Script” button, the new molecule (“vm”) should be added with all the settings you’ve given it:
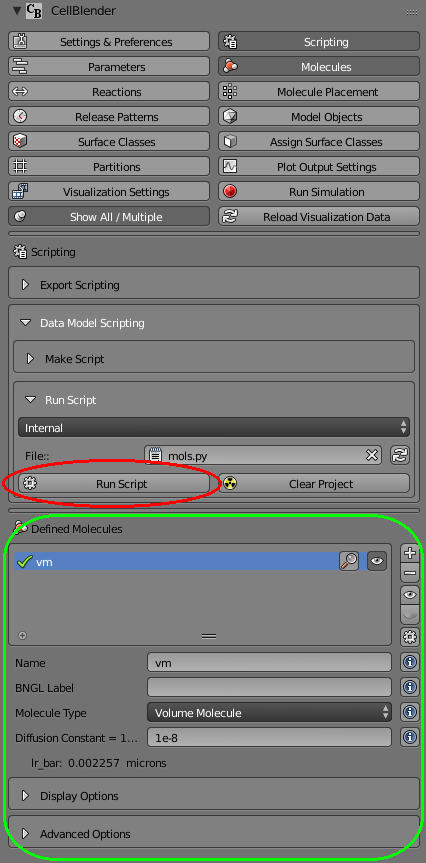
Congratulations, you’ve just written and executed your first data model script!
14.1.4.2.1. Discovering Data Model Fields¶
When we created our script, we had to “know” the names of the various fields in the Data Model. We had to know that there was an “mcell” field containing a “define_molecules” dictionary containing a “molecule_list” holding the individual molecules. We also had to know the fields inside the molecule (‘mol_name’, ‘mol_type’, and ‘diffusion_constant’). We also had to “know” the types of values that they took. Where is all of that information documented?
The easy answer is that it’s documented in the Data Model itself. We can build whatever we want through CellBlender’s normal panels (define molecules, reactions, release sites, etc) and then examine their data model representations to build our script.
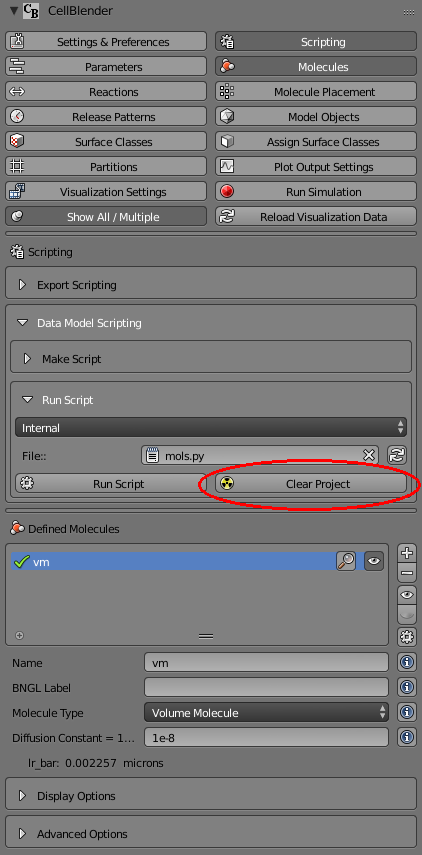
Start by pressing the “Clear Project” button. This button will completely remove all parts of your model (molecules, reactions, surface classes, etc):
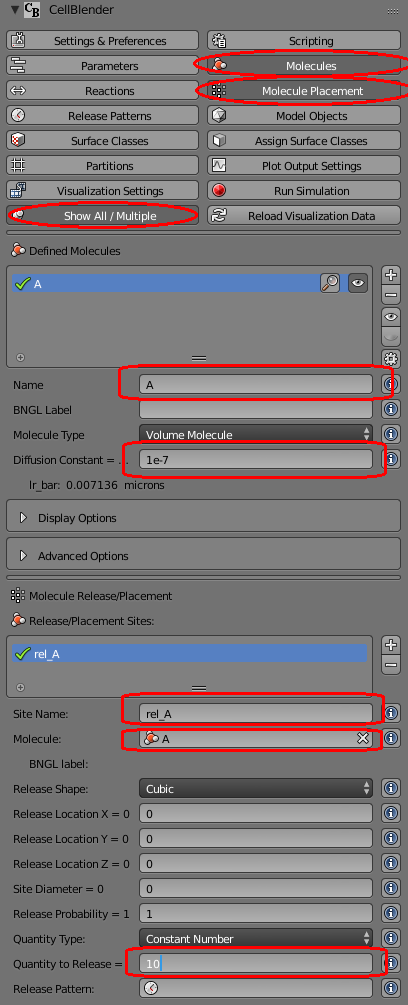
Now let’s build a simple model that we can explore through the data model. Our simple model will have one molecule type (named “A”) with a diffusion constant of 1e-7 and we will release 10 of them at the origin. Your panel should look like this (with the “Molecules” panel and “Molecule Placement” panel showing the settings):
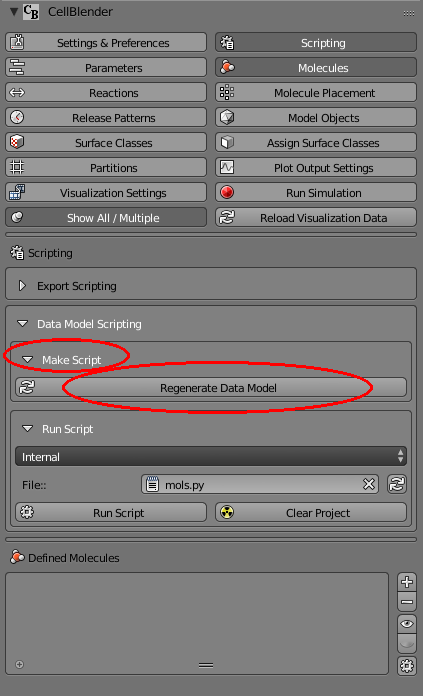
Then close the “Molecule Placement” panel and reopen the “Scripting” panel. Open the “Make Script” subpanel (inside “Data Model Scripting”) and click the “Regenerate Data Model” button:
That panel should then be showing a few new controls including a data model section selector (typically defaults to showing “Molecules”):
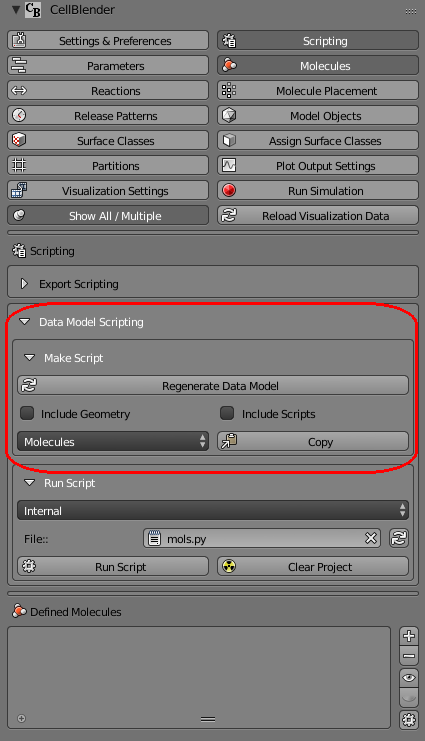
If you click on the “Molecules” control, you’ll see a pop up menu of all the different data model sections in your current model.
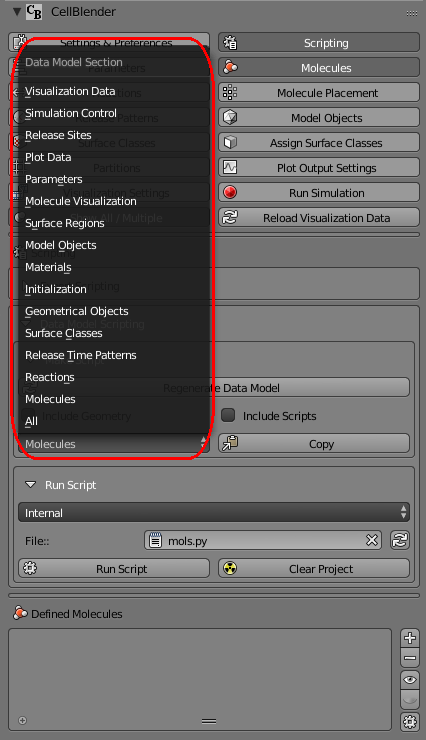
Select the “Molecules” section because that’s what we’d like to explore. Then click the “Copy” button. This will copy the entire “Molecules” section of the data model onto the clipboard.
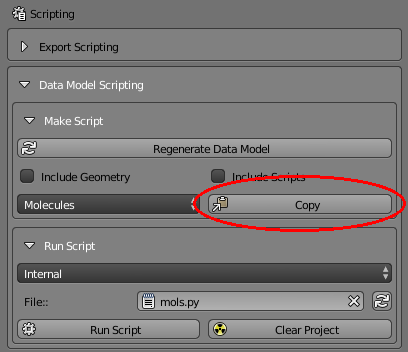
Now go back to the text editing window and position your cursor at the end (bottom) of the file (you might add a few carriage returns for spacing). Then paste (Control-V or Edit/Paste) the clipboard data into the text editor. You should see new text added (shown highlighted) in the text window:
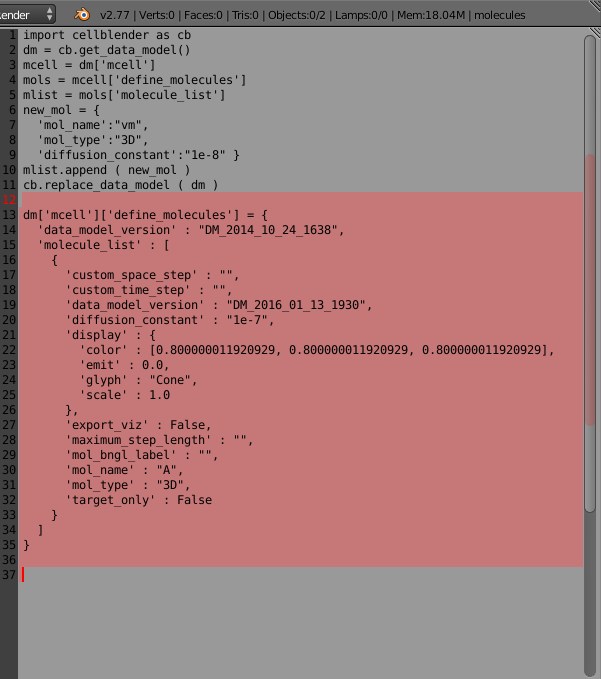
Here’s the new code that was added:
dm['mcell']['define_molecules'] = {
'data_model_version' : "DM_2014_10_24_1638",
'molecule_list' : [
{
'custom_space_step' : "",
'custom_time_step' : "",
'data_model_version' : "DM_2016_01_13_1930",
'diffusion_constant' : "1e-7",
'display' : {
'color' : [0.800000011920929, 0.800000011920929, 0.800000011920929],
'emit' : 0.0,
'glyph' : "Cone",
'scale' : 1.0
},
'export_viz' : False,
'maximum_step_length' : "",
'mol_bngl_label' : "",
'mol_name' : "A",
'mol_type' : "3D",
'target_only' : False
}
]
}
You’ll notice that it contains the same structure as before (an “mcell” dictionary containing a “define_molecules” dictionary containing a “molecule_list” list). And inside the molecule, you’ll see the familiar fields: ‘mol_name’ and ‘diffusion_constant’. But you’ll also see all of the other fields that were defined when you created your “A” molecule within the CellBlender interface. This provides an easy way to see how to program all of the settings within the CellBlender interface.
14.1.4.2.2. Looping to Release a Grid of Molecules¶
For our purposes in this tutorial, we want to create a script that will generate a grid of molecules with different diffusion constants in the x-y plane. For simplicity, we’ll set the diffusion constant proportional to the sum of the x and y coordinates. We’ll name each molecule according to its x and y locations. Assuming an 8x8 grid the outline of our code would look like this:
for x in range(8):
for y in range(8):
mol_name = "Grid_" + str(x) + "_" + str(y)
diffusion_constant = 1e-7 * (x + y)
# Create a molecule here
So what goes in the “Create a molecule here” section? It’s exactly what we got from the data model with the substitution of our calculated molecule names and diffusion constants.
Here’s what that code looks like:
import cellblender as cb
dm = cb.get_data_model()
mcell = dm['mcell']
mols = mcell['define_molecules']
mlist = mols['molecule_list']
for x in range(8):
for y in range(8):
mol_name = "Grid_" + str(x) + "_" + str(y)
diffusion_constant = 1e-7 * (x + y)
# Create a molecule here
new_mol = {
'custom_space_step' : "",
'custom_time_step' : "",
'data_model_version' : "DM_2016_01_13_1930",
'diffusion_constant' : str(diffusion_constant),
'display' : {
'color' : [0.8, 0.8, 0.8],
'emit' : 0.0,
'glyph' : "Cube",
'scale' : 1.0
},
'export_viz' : False,
'maximum_step_length' : "",
'mol_bngl_label' : "",
'mol_name' : mol_name,
'mol_type' : "3D",
'target_only' : False
}
mlist.append ( new_mol )
cb.replace_data_model ( dm )
You’ll notice that we rounded the colors from 0.80000… down to 0.8, and changed the glyph from the default “Cone” to “Cube”. We also substituted “mol_name” where we previously used “A”, and we replaced our fixed diffusion constant of “1e-7” with a string version of the diffusion constant that we calculated. We could tell it had to be a string because it was already a string in the data model that we copied from CellBlender.
We also moved the “cb.replace_data_model(dm)” call to the end (where it belongs). This gives us the general flow from top to bottom:
1. Import cellblender
2. Get the data model
3. Create convenience variables
4. Modify the data model lists and dictionaries
5. Replace the data model
Now we can clear the project, and run the script:
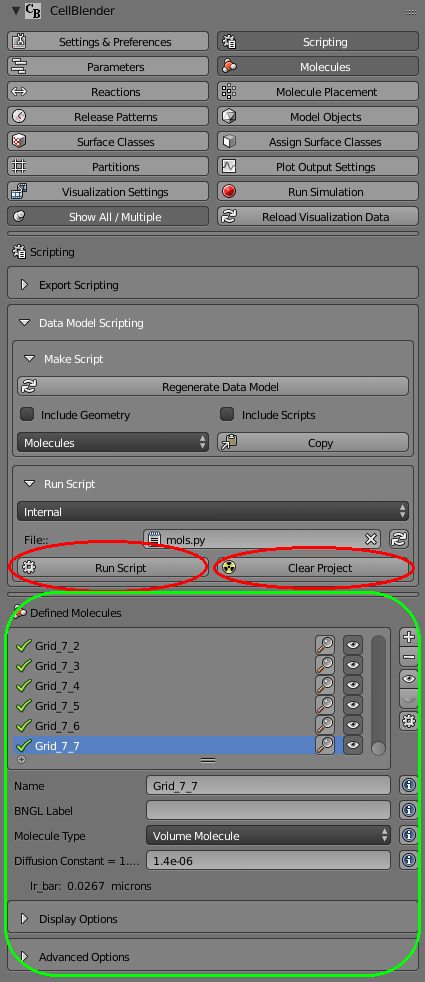
As shown, you should see a bunch of molecules that have been created with names ranging from “Grid_0_0” to “Grid_7_7”. If you browse through them, you’ll see diffusion constants ranging from 0.0 up to 1.4e-6.
Note that if you run the script a second time without pressing the “Clear Project” button, you will be attempting to create duplicate molecules, and you’ll see lots of “Duplicate Molecule” errors (try it and see). If that happens, just clear the project and run the script again. If this script was intended to add on to an existing model, it could check the data model first to see which molecules already existed before recreating them or build a dictionary of existing names and generate new names not in the dictionary. In our case, we’re not trying to add to an existing model, so we can just clear the existing project every time we run.
Now that we’ve defined all of our molecules, we need to release them at the coordinates of our grid. How do we find out how to script release sites? Just as with the molecules, we can create one in CellBlender, and then copy it to the clipboard and paste it into our script. Then we can modify it as needed.
Our panels are getting a little crowded, so release the “Show All / Multiple” push pin and open just the “Molecule Placement” panel. Then click the “+” button to add a new molecule release site:
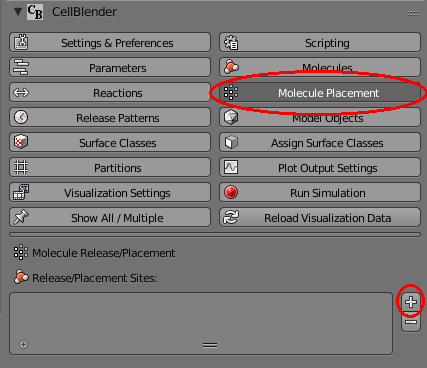
Change it to release molecule “Grid_0_0” and set the Quantity to 10:
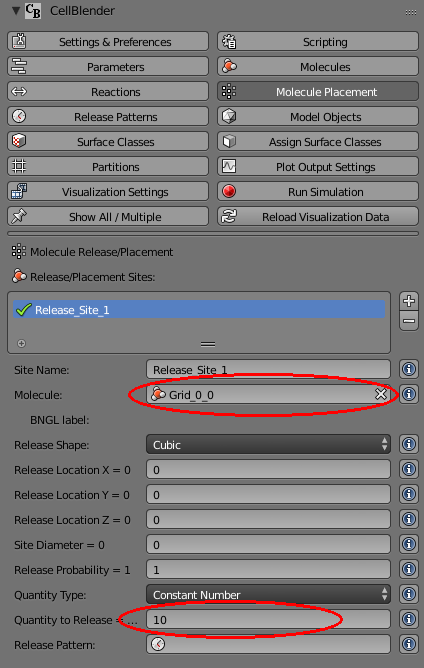
Now let’s copy the data model definitions from our new release site to the clipboard by opening the Scripting panel and choosing the “Release Sites” selection. Then click the copy button to make a copy on the clipboard:
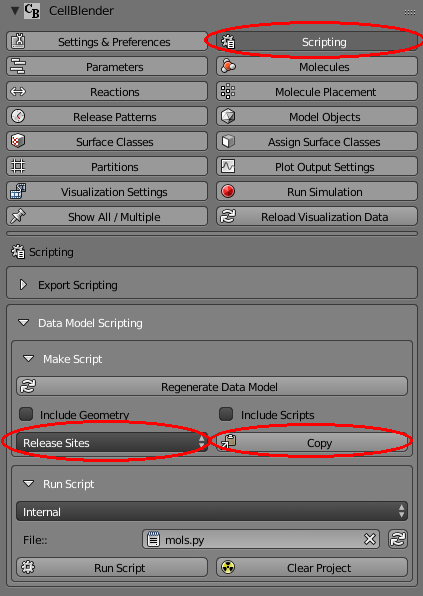
Then we can go to the bottom of our script and paste these release site definitions into our code (it’s good to add a few blank lines to separate the new section of text). Here’s what that looks like in the text editor with the new code highlighted:
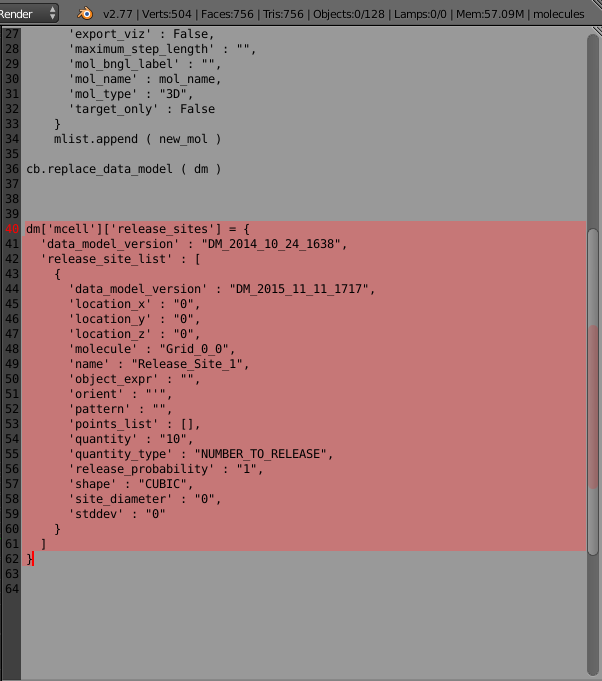
Here’s the new code that was just pasted:
dm['mcell']['release_sites'] = {
'data_model_version' : "DM_2014_10_24_1638",
'release_site_list' : [
{
'data_model_version' : "DM_2015_11_11_1717",
'location_x' : "0",
'location_y' : "0",
'location_z' : "0",
'molecule' : "Grid_0_0",
'name' : "Release_Site_1",
'object_expr' : "",
'orient' : "'",
'pattern' : "",
'points_list' : [],
'quantity' : "10",
'quantity_type' : "NUMBER_TO_RELEASE",
'release_probability' : "1",
'shape' : "CUBIC",
'site_diameter' : "0",
'stddev' : "0"
}
]
}
There are a number of ways to integrate this into our existing code. We could integrate it into the existing loop or add a second loop. In this case, we’ll integrate it into the existing loop. Here’s the code:
import cellblender as cb
dm = cb.get_data_model()
mcell = dm['mcell']
mols = mcell['define_molecules']
mlist = mols['molecule_list']
rels = mcell['release_sites']
rlist = rels['release_site_list']
for x in range(8):
for y in range(8):
mol_name = "Grid_" + str(x) + "_" + str(y)
diffusion_constant = 1e-7 * (x + y)
# Create a molecule here
new_mol = {
'custom_space_step' : "",
'custom_time_step' : "",
'data_model_version' : "DM_2016_01_13_1930",
'diffusion_constant' : str(diffusion_constant),
'display' : {
'color' : [0.8, 0.8, 0.8],
'emit' : 0.0,
'glyph' : "Cube",
'scale' : 1.0
},
'export_viz' : False,
'maximum_step_length' : "",
'mol_bngl_label' : "",
'mol_name' : mol_name,
'mol_type' : "3D",
'target_only' : False
}
mlist.append ( new_mol )
# Create a release site here
new_rel = {
'data_model_version' : "DM_2015_11_11_1717",
'location_x' : str(x),
'location_y' : str(y),
'location_z' : "0",
'molecule' : mol_name,
'name' : "Rel_" + mol_name,
'object_expr' : "",
'orient' : "'",
'pattern' : "",
'points_list' : [],
'quantity' : "10",
'quantity_type' : "NUMBER_TO_RELEASE",
'release_probability' : "1",
'shape' : "CUBIC",
'site_diameter' : "0",
'stddev' : "0"
}
rlist.append ( new_rel )
cb.replace_data_model ( dm )
As before, we can clear the project and run the script. We can also run the simulation and refresh the molecule display. Here’s what that looks like so far:
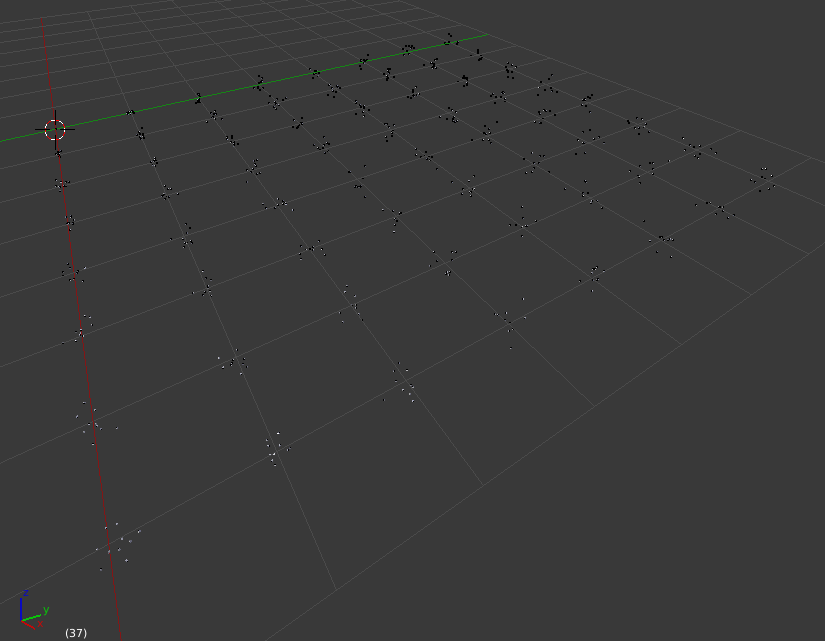
At this scale, the molecules are very small and virtually invisible. Let’s make them larger by going to the “Molecules” panel, and opening the “Display Options” for molecule “Grid_7_7”, and changing its Scale Factor from 1.0 to 10.0:
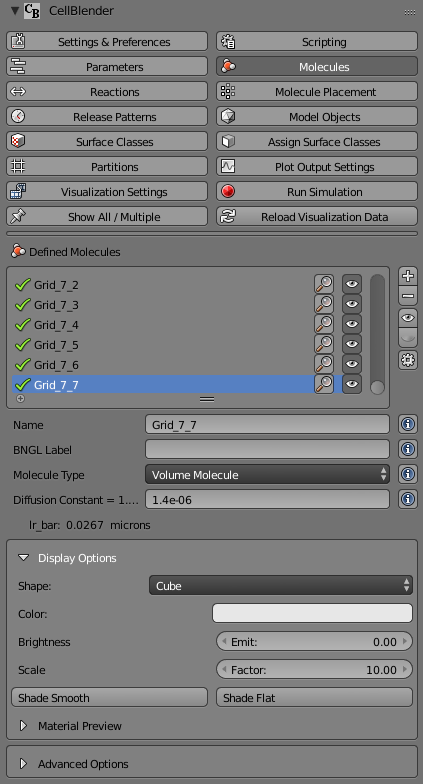
That makes them nice and visible:
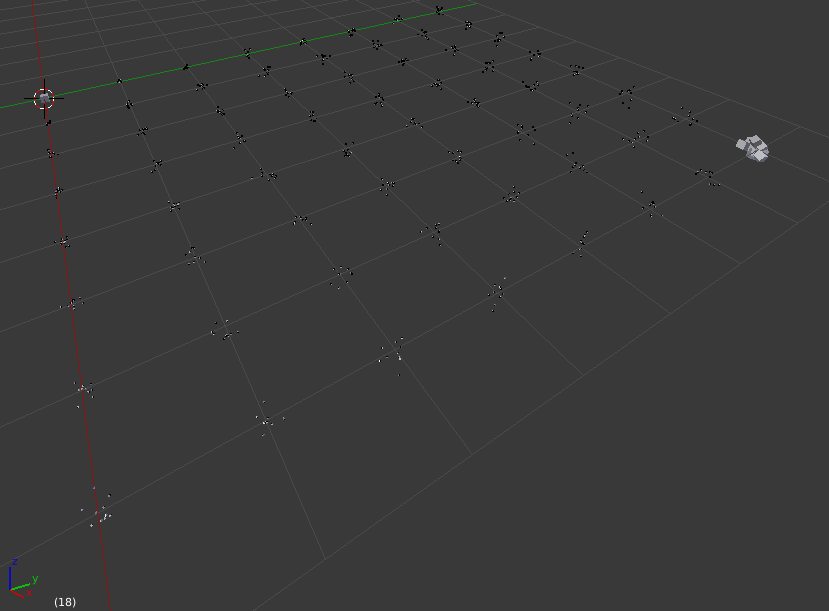
Now we can go back to our script and change the scale to 10:
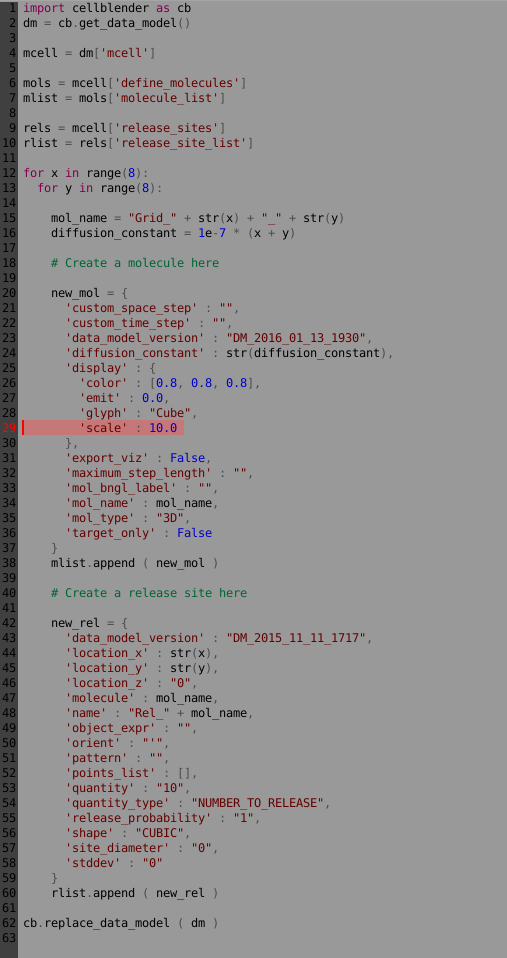
Clear the Project, then re-run the script (no need to re-reun the simulation). Refresh the molecules and it should look something like this:

For a “finishing touch” experiment with color by increasing the emit value, and changing the molecule color line to something like this:
'color' : [ (x+y)%3, (x%2), -((y%2)-1) ],
'emit' : 1.0,
14.1.4.2.3. Results and Final Script¶
If you run the simulation again and reload the visualization data it should look something like this:

Here’s the final script:
import cellblender as cb
dm = cb.get_data_model()
mcell = dm['mcell']
mols = mcell['define_molecules']
mlist = mols['molecule_list']
rels = mcell['release_sites']
rlist = rels['release_site_list']
for x in range(8):
for y in range(8):
mol_name = "Grid_" + str(x) + "_" + str(y)
diffusion_constant = 1e-7 * (x + y)
# Create a molecule here
new_mol = {
'custom_space_step' : "",
'custom_time_step' : "",
'data_model_version' : "DM_2016_01_13_1930",
'diffusion_constant' : str(diffusion_constant),
'display' : {
'color' : [(x+y)%3, (x%2), -((y%2)-1)],
'emit' : 1.0,
'glyph' : "Cube",
'scale' : 10.0
},
'export_viz' : False,
'maximum_step_length' : "",
'mol_bngl_label' : "",
'mol_name' : mol_name,
'mol_type' : "3D",
'target_only' : False
}
mlist.append ( new_mol )
# Create a release site here
new_rel = {
'data_model_version' : "DM_2015_11_11_1717",
'location_x' : str(x),
'location_y' : str(y),
'location_z' : "0",
'molecule' : mol_name,
'name' : "Rel_" + mol_name,
'object_expr' : "",
'orient' : "'",
'pattern' : "",
'points_list' : [],
'quantity' : "10",
'quantity_type' : "NUMBER_TO_RELEASE",
'release_probability' : "1",
'shape' : "CUBIC",
'site_diameter' : "0",
'stddev' : "0"
}
rlist.append ( new_rel )
cb.replace_data_model ( dm )